Modelering van luminescente materialen via moleculaire dynamica van geexciteerde toestanden
Modelering van luminescente materialen via moleculaire dynamica van geexciteerde toestanden
Promotor(en): K. Hemelsoet /MM_13_SPEC04 / Model and software development, SpectroscopyLuminescente materialen zenden zichtbaar licht uit door de overgang van een hoog- naar een laag-energetische toestand. Zij worden in uiteenlopende toepassingsdomeinen gebruikt. Klassieke toepassingsgebieden zijn de verlichting- en beeldschermindustrie, maar recent worden ook nieuwe materiaaltypes met luminescente eigenschappen onderzocht en geproduceerd en is er veel aandacht voor nieuwe toepassingsgebieden, waaronder sensoren, zonnecellen of om geneesmiddelen te traceren.
Om luminescente eigenschappen van moleculen en materialen goed te begrijpen, is een gedetailleerde kennis van de structuur noodzakelijk. Met moet het potentiële energie-oppervlak (potential energy surface, PES) van zowel de grondtoestand (aangeduid met S0 in de figuur) als van geëxciteerde toestanden (S1 en S2 in de figuur) zo accuraat mogelijk beschrijven. De theoretische modellering van deze PES oppervlakken is zeer uitdagend, en dit in het bijzonder voor de geëxciteerde toestanden.
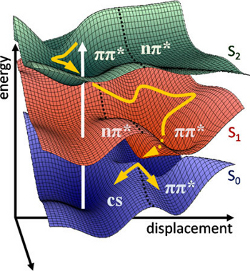 In het Centrum voor Moleculaire Modelering wordt er veel onderzoek uitgevoerd naar de accurate berekening van verschillende elektronische spectra. Bij de berekening van lichtabsorptie (dus de overgang van de grondtoestand naar een geëxciteerde toestand) bleek dat een statische benadering te kort schiet om accurate voorspellingen te doen. Een statische simulatie houdt in dat er gezocht wordt naar een energetisch gunstige geometrie op een welbepaald tijdstip. Een realistisch veeldeeltjessysteem is echter dynamisch en varieert in de tijd, en met behulp van Moleculaire Dynamica (MD) simulaties kan de tijdsevolutie van de moleculaire geometrie in rekening gebracht worden. Dit levert een ensemble van geometrieën op. Voor de bepaling van absorptie-energieën is een uitmiddeling over dit ensemble een zeer succesvolle methode gebleken.
In het Centrum voor Moleculaire Modelering wordt er veel onderzoek uitgevoerd naar de accurate berekening van verschillende elektronische spectra. Bij de berekening van lichtabsorptie (dus de overgang van de grondtoestand naar een geëxciteerde toestand) bleek dat een statische benadering te kort schiet om accurate voorspellingen te doen. Een statische simulatie houdt in dat er gezocht wordt naar een energetisch gunstige geometrie op een welbepaald tijdstip. Een realistisch veeldeeltjessysteem is echter dynamisch en varieert in de tijd, en met behulp van Moleculaire Dynamica (MD) simulaties kan de tijdsevolutie van de moleculaire geometrie in rekening gebracht worden. Dit levert een ensemble van geometrieën op. Voor de bepaling van absorptie-energieën is een uitmiddeling over dit ensemble een zeer succesvolle methode gebleken.Indien men gelijkaardige methoden wil toepassen om luminescentie te beschrijven, is echter een ensemble van moleculaire geometrieën in de geëxciteerde toestand nodig. Hiervoor is een accurate beschrijving van de geëxciteerde potentiële energie-oppervlakken (S1 of S2) nodig.
Het doel van deze thesis is om Moleculaire Dynamica simulaties op deze geëxciteerde energie-oppervlakken uit te voeren. Dit kan aan de hand van kwantummechanische technieken (Car-Parinello Molecular Dynamics, CPMD) of door het opstellen van een Newtoniaans krachtveld. In eerste instantie zullen kleinere modelsystemen bestudeerd worden, waarop de verschillende methoden uitvoerig worden getest, om vervolgens toe te passen op grotere systemen. Afhankelijk van de specifieke interesse van de geїnteresseerde student zal er meer aandacht gegeven worden aan de modelontwikkeling en –evaluatie of aan de toepassingen. Er is geen voorkennis vereist van kwantumfysische pakketten, dit wordt tijdens het thesisjaar aangeleerd.
 In het Centrum voor Moleculaire Modelering wordt er veel onderzoek uitgevoerd naar de accurate berekening van verschillende elektronische spectra. Bij de berekening van lichtabsorptie (dus de overgang van de grondtoestand naar een geëxciteerde toestand) bleek dat een statische benadering te kort schiet om accurate voorspellingen te doen. Een statische simulatie houdt in dat er gezocht wordt naar een energetisch gunstige geometrie op een welbepaald tijdstip. Een realistisch veeldeeltjessysteem is echter dynamisch en varieert in de tijd, en met behulp van Moleculaire Dynamica (MD) simulaties kan de tijdsevolutie van de moleculaire geometrie in rekening gebracht worden. Dit levert een ensemble van geometrieën op. Voor de bepaling van absorptie-energieën is een uitmiddeling over dit ensemble een zeer succesvolle methode gebleken.
In het Centrum voor Moleculaire Modelering wordt er veel onderzoek uitgevoerd naar de accurate berekening van verschillende elektronische spectra. Bij de berekening van lichtabsorptie (dus de overgang van de grondtoestand naar een geëxciteerde toestand) bleek dat een statische benadering te kort schiet om accurate voorspellingen te doen. Een statische simulatie houdt in dat er gezocht wordt naar een energetisch gunstige geometrie op een welbepaald tijdstip. Een realistisch veeldeeltjessysteem is echter dynamisch en varieert in de tijd, en met behulp van Moleculaire Dynamica (MD) simulaties kan de tijdsevolutie van de moleculaire geometrie in rekening gebracht worden. Dit levert een ensemble van geometrieën op. Voor de bepaling van absorptie-energieën is een uitmiddeling over dit ensemble een zeer succesvolle methode gebleken.- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsSpectroscopie, Moleculaire Modellering, Optische eigenschappen, Moleculaire dynamicaRecommended coursesSimulations and modeling for the nanoscale (EA); Veeldeeltjesfysica (WE); Computationele Fysica (WE)
Contact
Prof. Dr. ir. Karen Hemelsoet
Louis Vanduyfhuys