Machine learning of the ground state density of molecules for force field development
Machine learning of the ground state density of molecules for force field development
Promotor(en): T. Verstraelen /25298 / Model and software developmentBackground and problem
Computational simulations have for decades become an invaluable tool to gain a fundamental understanding of the properties of materials at the atomic scale. To study materials containing a large number of atoms, one often relies on force fields, which are composed of analytical functions describing the interatomic interactions. These force fields are preferably derived from ab-initio calculations, as no experimental input is required in this case. Recently, an intermolecular force field was published in our group which requires as input only the ab-initio electron density of the molecule(s) under study.[1] Although such ab-initio calculations are relatively cheap for small molecules, the computational cost quickly increases with increasing system size. This prevents the derivation of accurate force fields for very large molecules or periodic structures. Therefore, a computationally efficient method to obtain a partitioning of the electron density into atomic fragments of large systems is a very relevant avenue for research. We propose the use of neural networks for the prediction of the partitioned density, solely from the input of the geometry of the molecule (see Figure 1).
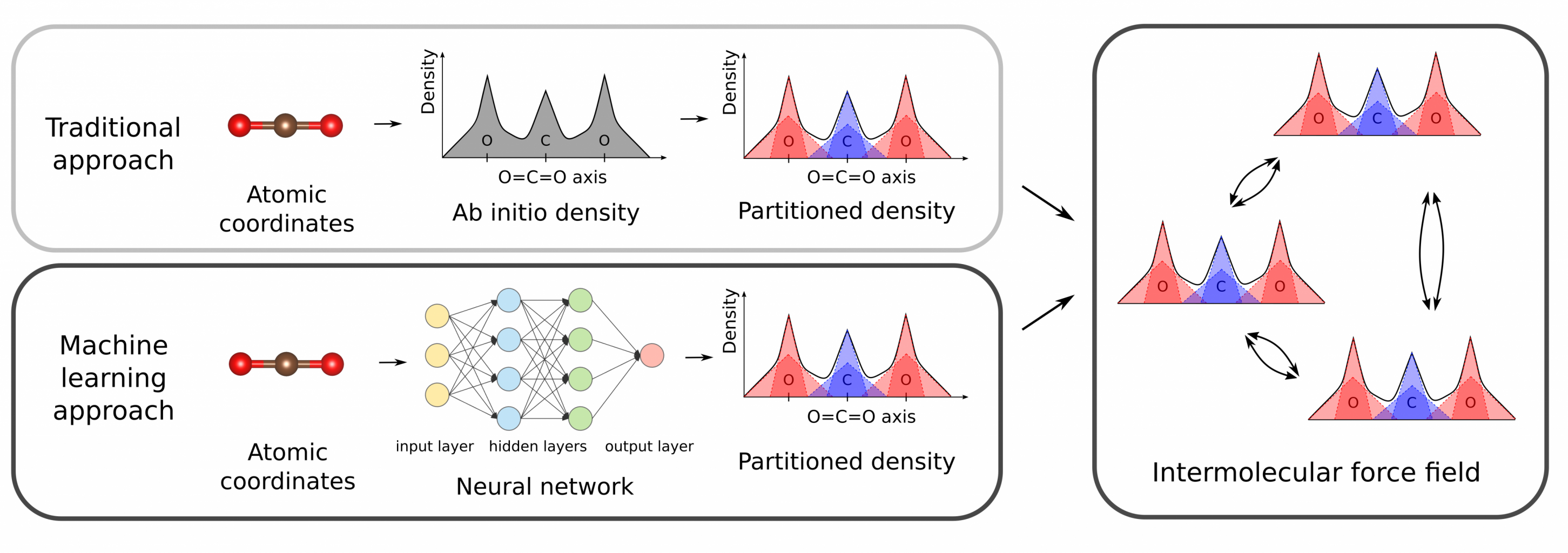
Figure 1. Traditional and machine learning approach to derive an atomic density partitioning for intermolecular force fields.
Goal
The first step in this thesis is a critical evaluation of an existing kernel-based approach from the literature.[2] Its performance on a challenging set of zwitterionic structures will be assessed. The model is limited to neutral molecules containing only C, N, H and O atoms, and can not be used to construct force fields for ionic molecules and liquids.
The second objective is therefore to construct a neural network model, based on an existing architecture called SchNet. [3] This network was originally designed to predict the energy of a molecule, and it will therefore be modified to yield the partitioned densities of the molecule. To train the model, the ab-initio density of molecules in the existing QM9 dataset[4], as well as ionic molecules from several datasets will be calculated. The prediction error on the total and partitioned densities will be evaluated, and if not satisfactory, further modifications will be made to the network.
In a second step, the predicted densities will be benchmarked further by calculating the electrostatic interaction between dimers from a dataset of nucleobase pairs. This will serve as a validation of the model in yielding accurate electrostatic force fields without the need for expensive ab-initio calculations.
Lastly, a more challenging task is to predict the partitioned density of porous metal-organic frameworks, for which the CoRE MOF database will serve as training data. [5] These MOFs are promising candidates for future adsorption applications, and are therefore often investigated in computational high-throughput screenings of adsorption properties. However, these studies are often limited by the availability of atomic charges; a limitation which could be alleviated by a trained ML network.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]Keywordsmachine learning, Computational physics, density partitioning, Force fieldsReferences
[1] S. Vandenbrande, M. Waroquier, V. Van Speybroeck & T. Verstraelen (2017) The Monomer Electron Density Force Field (MEDFF): A Physically Inspired Model for Noncovalent Interactions. Journal of Chemical Theory and Computation, 13(1), 161–179. https://doi.org/10.1021/acs.jctc.6b00969
[2] T. Bereau, R. A. DiStasio Jr., A. Tkatchenko, O. A. Lilienfeld (2018) Non-covalent interactions across organic and biological subsets of chemical space: Physics-based potentials parametrized from machine learning. The Journal of Chemical Physics, 148, 241706. https://doi.org/10.1063/1.5009502
[3] K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko & K.-R. Müller (2017) SchNet: SchNet – A deep learning architecture for molecules and materials. The Journal of Chemical Physics, 148(24). https://doi.org/10.1063/1.5019779
[4] R. Ramakrishnan, P. O. Dral, M. Rupp, O. A. von Lilienfeld, Quantum chemistry structures and properties of 134 kilo molecules, Scientific Data 1, 140022, 2014 https://doi.org/10.1038/sdata.2014.22
[5] Y.G. Chung et al., Advances, Updates, and Analytics for the Computation-Ready, Experimental Metal–Organic Framework Database: CoRE MOF 2019, Journal of Chemical & Engineering Data, 64(12), 2019 https://doi.org/10.1021/acs.jced.9b00835
Contact
Toon Verstraelen