Investigating adsorption-induced flexibility in metal-organic frameworks using machine learning potentials
Investigating adsorption-induced flexibility in metal-organic frameworks using machine learning potentials
Promotor(en): V. Van Speybroeck, T. Verstraelen /20MODEV09 / Model and software developmentProbleemstelling:
Over the past three decades, metal-organic frameworks (MOFs) have emerged as an exceedingly promising class of nanoporous materials due to their extraordinary behaviour and endless functional variety. They are composed of inorganic nodes and organic ligands, joined together with coordination bonds to form two- and three-dimensional networks. The vast number of possibilities in network topology and chemical composition of the organic and inorganic building blocks has given rise to a number of fascinating physical properties, such as their exceptionally high specific surface areas or the ability to undergo structural phase transitions induced by external stimuli such as breathing. These properties give MOFs much potential for a wide variety of applications that involve either the separation or temporary storage of specific gases of interest such as methane, carbon dioxide and water. As a result, enormous experimental and computational research efforts have been dedicated to the design and synthesis of MOFs exhibiting targeted adsorption or stimuli-responsive behaviour for use in specific applications. Computational modeling at the molecular level is a particularly attractive approach as it allows to predict which MOFs are best suited for which applications without the need to perform a large number of expensive experiments. Unfortunately, traditional computational methodologies often fail when it comes to an accurate description of flexibility as induced by the adsorption of certain guest molecules such as carbon dioxide or water. While adsorption-induced flexibility is an intrinsically dynamic phenomenon that requires advanced molecular simulations on relatively large timescales, it is important to note that the interaction of polarizable molecules such as water or carbon dioxide with the metal site in the inorganic node is extremely challenging to model. On one hand, highly accurate ab initio methods (such as density functional theory) are prohibitively expensive to use in long simulations, while computationally faster methods such as classical force fields simply do not correctly model this framework-guest interaction.
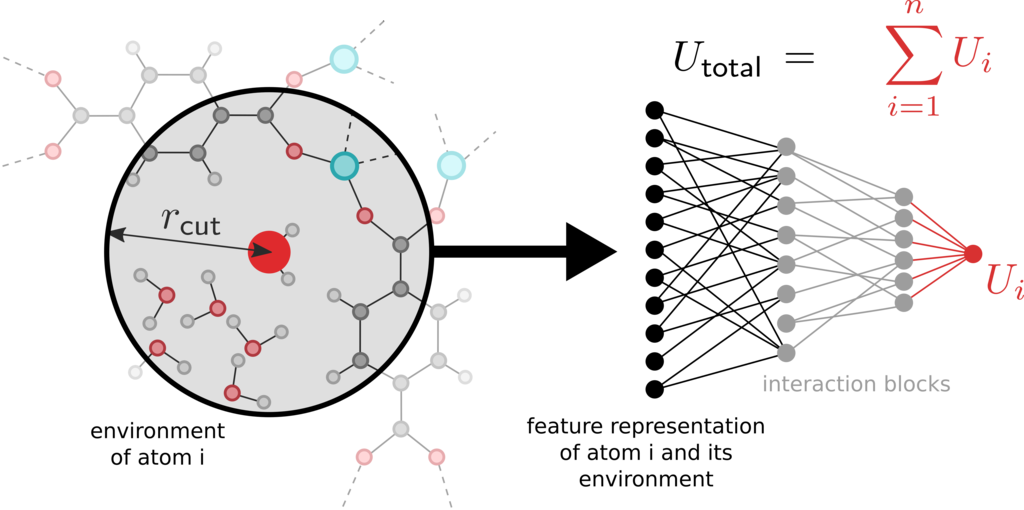
Doelstelling:
Here, we aim to develop a methodology that combines the accuracy of expensive ab initio calculations with the timescale available to more approximate methods such as force fields. A particularly promising approach – which has proven successful for other molecular systems – is to construct a machine learning model that learns the complex framework-guest interactions, but is fast enough to evaluate such that it can be employed in long, dynamic simulations. A variety of such machine learning potentials (MLPs) have been proposed [1, 2], and their parameterization relies on a large dataset of ab initio calculations of the total potential energy and the forces on each atom. However, prior work on their application to MOFs is sparse. Only one such model exists [3], but it has only been tested on a single rigid MOF and, more importantly, it does not include any framework-guest interactions. Our approach here will exploit the specific structure of MOFs to develop accurate machine learning models that are faster and cheaper to parameterize than more generic models. Specifically, we aim to reduce the model complexity and speed up training by additionally introducing a number of constraints based on physical insight. For example, the long-range limit of the model may be constrained to purely electrostatic interactions, or the framework-guest interaction may be treated separately from framework-framework interactions. By explicitly imposing such constraints, the network complexity may be reduced without a significant loss in accuracy. To develop the MLPs, we choose DMOF-1, a prototypical flexible MOF, in combination with carbon dioxide or benzene as our benchmark system [4]. First, several existing MLPs will be implemented and validated with their prediction of important physical quantities, such as (i) bond lengths and angles, (ii) pressure-versus-volume equation of state for the empty lattice, and (iii) adsorption isotherms. The insights learned from this analysis will facilitate the design of a new MLP that can go beyond state-of-the-art. After a rigorous validation of the developed model, we will use it in advanced simulation schemes such as the pressure-induced amorphization of DMOF-1 [5]. This complicated phenomenon cannot be described with conventional force fields, showing the added value of MLPs. Additionally, gas adsorption can be investigated in the more complex systems MIL-53(Al) [6] or CoBDP with the experience gained in studying DMOF-1.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]Keywordsmachine learning, molecular simulations, Nanoporous materialsReferences
[1] K. T. Schütt et al. J. Chem. Phys. 148, 241722 (2018) https://doi.org/10.1063/1.5019779
[2] A. S. Christensen et al. J. Chem. Phys. 152, 044107 (2020) https://doi.org/10.1063/1.5126701
[3] M. Eckhoff et al. J. Chem. Theory Comput. 15, 3793-3809 (2019) https://doi.org/10.1021/acs.jctc.8b01288
[4] L. Vanduyfhuys et al. Nat. Commun. 9, 204 (2018) https://doi.org/10.1038/s41467-017-02666-y
[5] J. Wieme et al. J. Mater. Chem. 7, 22663 (2019) https://doi.org/10.1039/C9TA01586H
[6] S.M.J. Rogge et al. Adv. Theory Simul. 2, 1800177 (2019) https://doi.org/10.1002/adts.201800177
Contact
Veronique Van Speybroeck
Toon Verstraelen