Exploring the black-to-yellow phase transformation mechanism of metal halide perovskites via machine learning potentials
Exploring the black-to-yellow phase transformation mechanism of metal halide perovskites via machine learning potentials
Promotor(en): V. Van Speybroeck, T. Verstraelen /20NANO11 / Nanoporous materialsProbleemstelling:
While the photovoltaic industry has always been dominated by silicon technology, the production of crystalline silicon is difficult and expensive. As a result, recent research efforts are dedicated towards new materials for use in cheaper solar cells. One of the most promising classes of candidate materials are perovskites. They garnered intensive interest because of their extraordinary properties for photovoltaic applications, such as as high absorptivity, long electron-hole diffusion lengths and high charge mobility. Recently, the maximum obtained solar cell efficiency of perovskite absorbers has risen substantially, up to a maximum of 25.2%, thereby rapidly closing the gap with more traditional photovoltaic materials. Perovskites are cheap due to their easy synthesis and have tuneable properties making them also interesting for other applications such as LEDs and X-ray detectors.
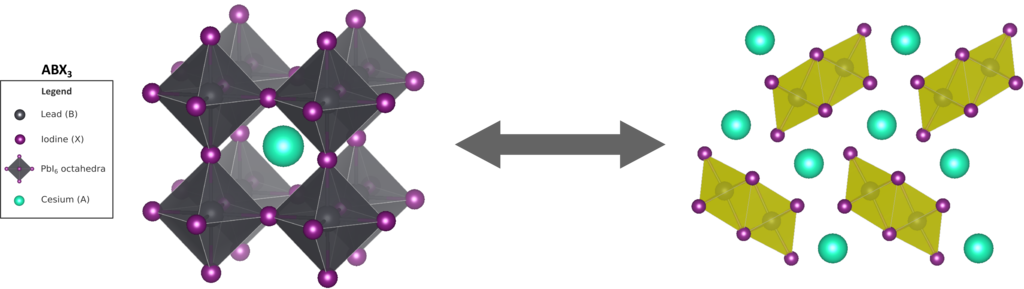
All perovskites have the same perovskite crystal structure, are described by an ABX3 chemical formula, and have a unit cell that is typically pseudo-cubic. The B cations are placed in the corners of the unit cell, the X anions in the center of the edges of the unit cell forming BX6 octahedra, and finally the A cations are placed in the center of the unit cell filling up the space in between the octahedra. For opto-electronic applications, we are interested in a subclass of perovskites, namely metal halide perovskites (MHPs), which have a metal atom for the B cation and a halide atom for the X anion. For MHPs, the major hurdle to overcome in going from research subject to commercial product is their limited mechanical and chemical stability. For example the most prominent MHP, methylammonium (MA) lead iodide (MAPbI3), degrades very fast in humid environments. While replacing the volatile MA cation with Cs improves the moisture stability significantly, other issues arise such as the polymorphic nature of CsPbI3. Experiments reveal that the desired black perovskite phase of CsPbI3 is metastable and tends to transition to a yellow nonperovskite phase, which is stable and associated with a complete loss of the desired opto-electronic properties. To prevent this degradation mechanism, the Center for Molecular Modeling (CMM) collaborated with the Hofkens group (KULeuven) to propose a new paradigm based on interfacial strain engineering [1]. To push the long-term stability of the black phase even further, it is necessary to understand how this methods stabilizes CsPbI3. Therefore it is crucial to acquire molecular-level insight into the physical interactions underpinning the stability of the material, and the mechanism through which the black-to-yellow phase transformation takes place.
Doelstelling:
In computer simulations, phase transformations are typically rare events that do not occur spontaneously. This complicates their investigation considerably as the attainable time scale in such simulations is often too small to observe a transition. In addition, the length scale in these simulations (i.e. the size of the periodic box) needs to be sufficiently large as periodic boundary conditions have been shown to introduce unphysical artefacts in the phase behavior of many systems [2]. Therefore, to study phase transitions in computer simulations, one often resorts to so-called enhanced sampling techniques which actively push the system from one phase to the other, thereby accelerating the time-to-transition significantly [3]. However, even with these enhanced sampling techniques, the computation time required to fully characterize the transition is orders of magnitude larger than what can be achieved by the current state of the art computing infrastructure. This is a consequence of the fact that these simulations are performed using a full quantum mechanical treatment of the interactions within the perovskites, which is prohibitively expensive to perform on large time and length scales. At this point, a traditional approach would approximate the quantum mechanical interactions with classical analytic expressions (in order to ensure a faster evaluation). Unfortunately, this is not possible for the black-to-yellow transformation discussed above, as it involves the breaking of multiple bonds, which cannot be captured by classical approximations to the quantum mechanical interactions. Therefore, we need to develop a new approximation with an accuracy comparable to ab initio methods, but at only a fraction of their computational cost.
In this thesis, we will tackle this problem by designing and applying a machine learning model that can predict the quantum mechanical energy and forces based on the atomic positions of the system (see Figure 2). By doing so, we can extend the time and length scale of the perovskite simulations by multiple orders of magnitude, which will enable us to gain fundamental new insight in the phase transformation discussed above. These machine learning potentials (MLPs) have been proven successful in a wide variety of molecular systems, including perovskites [4]. However, they have not yet been employed in studies on phase transformations, such as the black-to-yellow transformation discussed above.
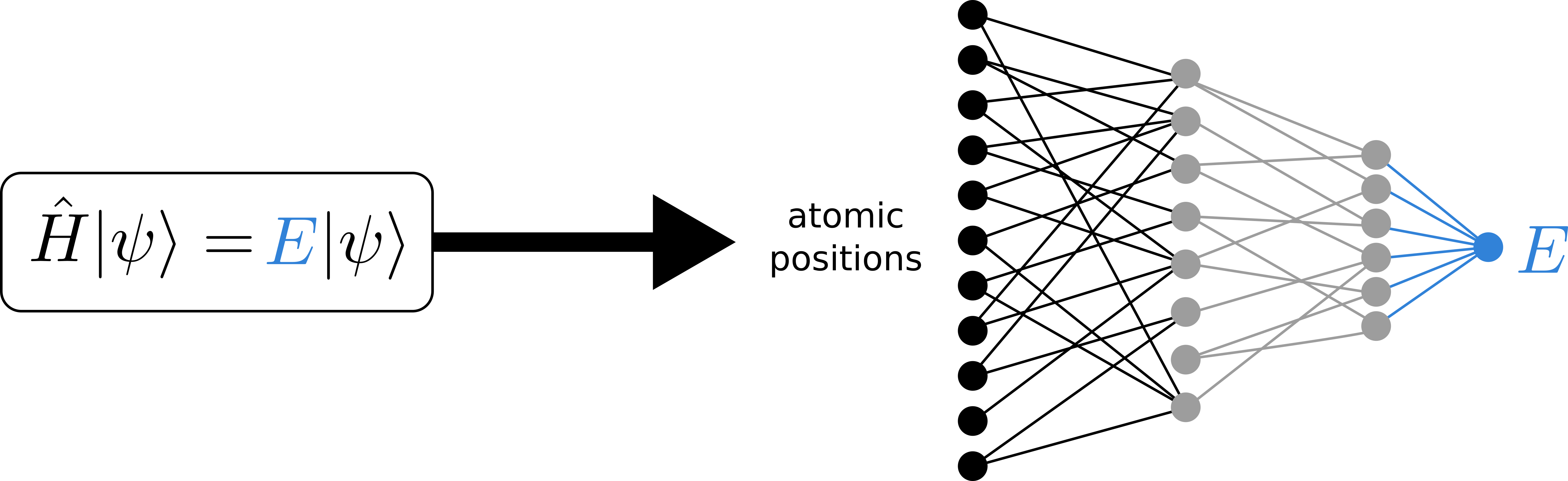
First, the student will get acquainted with the existing approaches to MLPs in perovskites by parameterizing and benchmarking these existing approaches on the black and yellow phases separately. In particular, this includes the deep-learning based SchNet architecture [5], to be trained based on a large dataset of quantum mechanical reference data that is already available at the CMM. In addition, we will explore a recently proposed kernel regression model that instead is trained on-the-fly, i.e. during a quantum mechanical reference simulation [4]. After the initial benchmarking of the MLPs in each phase separately, we aim to develop a procedure that can construct an MLP that is able to describe both phases at once. This model will then be used in combination with enhanced sampling techniques to study the black-to-yellow transformation on a much larger time and length scale than previously possible.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsPerovskites, machine learning, photovoltaics, free energy methods, nanoscale modelingReferences
[1] Steele et al., Science 365, 679-684 (2019)
[2] Rogge et al., Nat. Commun. 10, 4842 (2019)
[3] Demuynck et al., J. Chem. Theory Comput. 14, 5511-5526 (2018)
[4] Jinnouchi et al., Phys. Rev. Lett. 122, 225701 (2019)
[5] Schütt et al., J. Chem. Phys 148, 241722 (2018)
Contact
Veronique Van Speybroeck
Toon Verstraelen