Dynamic nano-sponges: nuclear quantum effects meet neural network potentials
Dynamic nano-sponges: nuclear quantum effects meet neural network potentials
Promotor(en): V. Van Speybroeck /28207 / Model and software development, Nanoporous materialsBackground and problem
Metal-organic frameworks (MOFs) are widely investigated because of their ability to capture and release water. In 2021, a research group from the University of Berkeley has patented a device to harvest atmospheric water in desert areas using MOFs. Much like an actual sponge, water is loaded or absorbed into frameworks through a network of small pores, and it may be unloaded using an external trigger such as mechanical pressure (squeezing the sponge) or temperature (heating the sponge) [1, 2]. Much unlike an actual sponge, the pore diameters of framework materials (1 nm) are orders of magnitude smaller than those found in everyday polymer sponges (100 µm). This difference in pore size distribution has incredibly large consequences for the behavior of water molecules. In particular, the water ‘droplets’ or clusters inside these nanometer-sized pores only contain about 10-100 molecules, and these will self-organize into a complex network of hydrogen bonds that depends on the shape and chemical properties of the surrounding pore (Figure 1). Interestingly, these clusters may exhibit different properties from ‘bulk’ water (e.g. in terms of density or coordination number). In addition, their presence in framework pores may actively modulate the mechanical stability of these frameworks because of specific interactions with framework atoms [3].
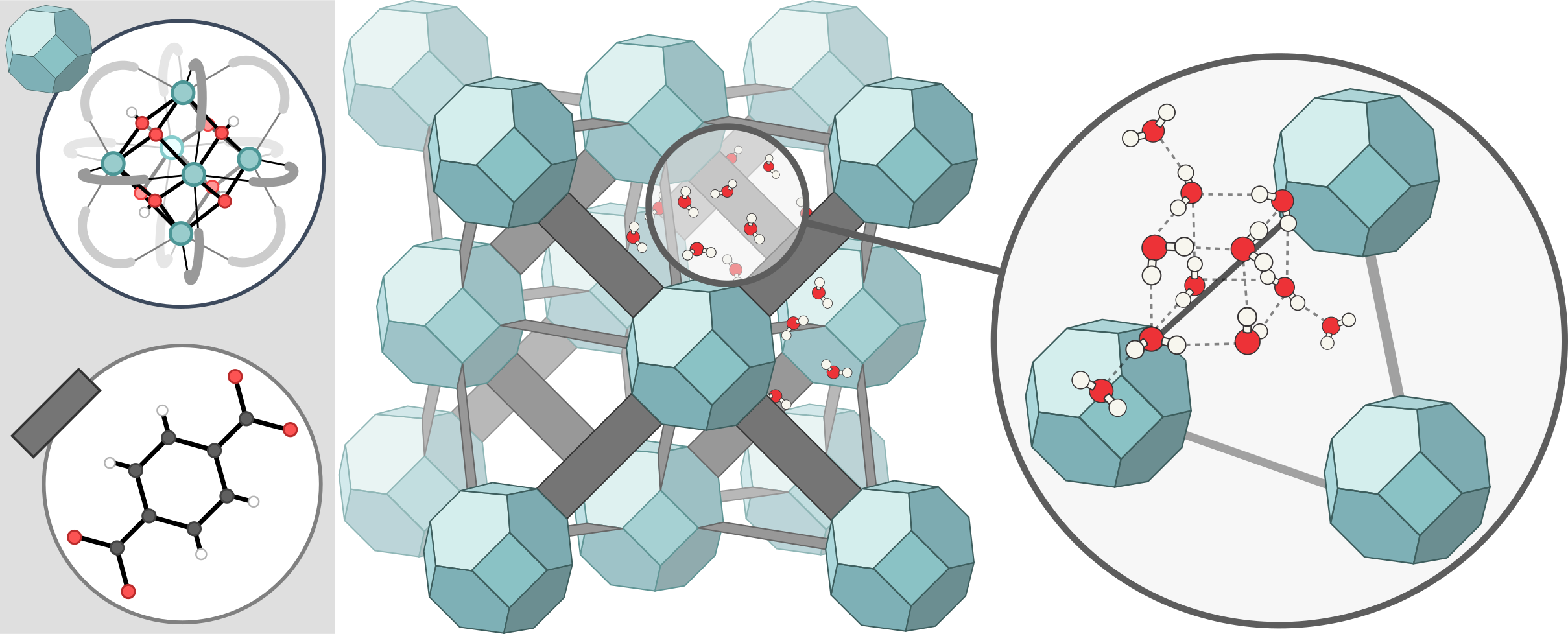
Figure 1: Schematic representation of UiO-66(Zr) and an associated cluster of water molecules.
To further develop MOFs into effective nano-sponges, it is necessary to obtain a clear understanding of the dynamics of water clusters and how they affect framework stability. Explicit atomistic simulation of water-loaded frameworks may aid in achieving this goal, and would additionally allow us to investigate the entire loading-unloading cycle. However, such simulations are extremely challenging for at least two reasons. First, hydrogen bonding is only well described if interatomic interactions are evaluated at the quantum mechanical level, i.e. by explicitly solving the Schrödinger equation to calculate the electron density throughout the system. Secondly, protons in water molecules are so small that their behavior is no longer well described by a classical point mass, and instead their nuclear wavefunction needs to be taken into account. These computational challenges have long hindered systematic investigations of the behavior of water-loaded frameworks. However, state-of-the-art simulation methods have now succeeded in replacing the expensive electron density calculation with a much more efficient neural network approximation of the interatomic interactions. This machine learning approach to quantum chemical simulations increases the computational efficiency by more than three orders of magnitude (Figure 2) [4].
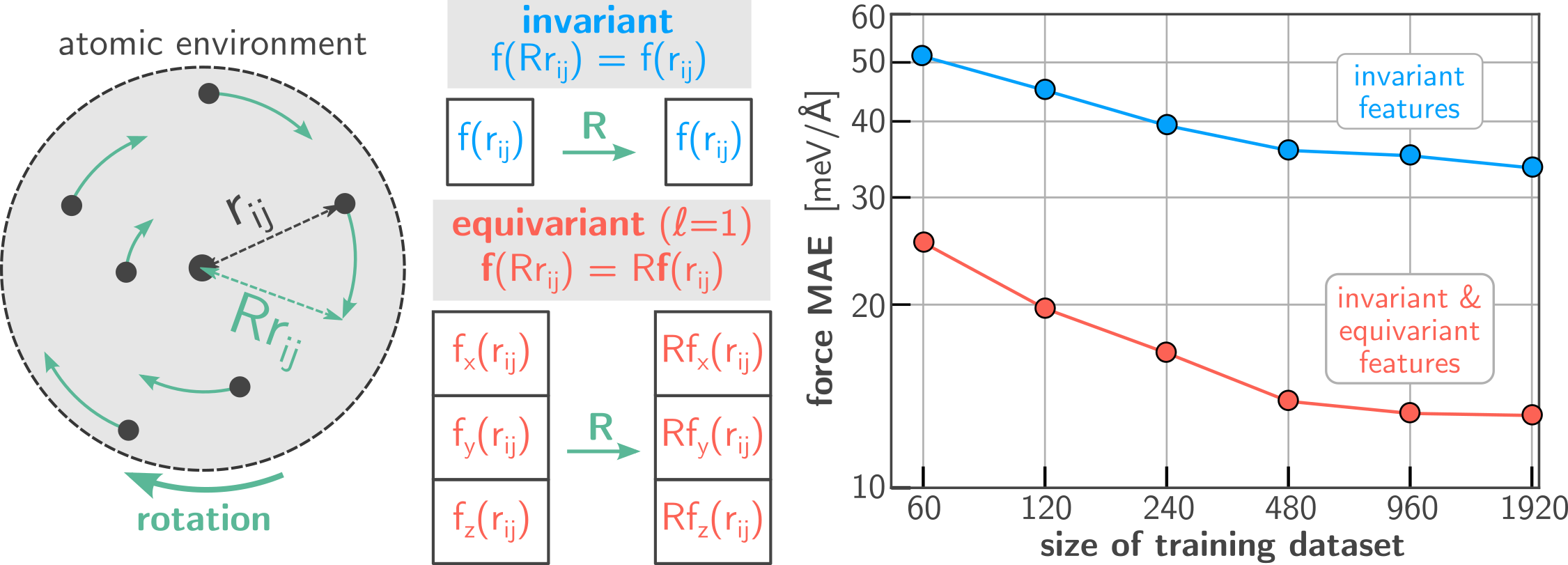
Figure 2: Illustration of rotationally equivariant feature representations of atomic environments (left). The accuracy of network potentials is greatly improved when intermediary feature representations are ‘directional’ in nature, allowing for a better characterization of the atomic environment. Neural network predictions are compared with density functional theory (DFT) reference data (right).
Goal
In this thesis, we aim to exploit the recent advances in neural network potentials to investigate the behavior of water in frameworks with an accuracy that is unprecedented in the field. To achieve this, we will build upon existing proof-of-concept work in which we successfully reproduced interatomic interactions in empty frameworks; the next step is to extend these results towards framework-guest interactions. This is nontrivial because intermolecular forces are up to an order of magnitude smaller than the covalent interactions between framework atoms, and may therefore be harder to learn. The student is encouraged to experiment with mixed-model descriptions, in which the total interaction energy is decomposed into framework-framework and framework-guest contributions, each of which described by its own interaction potential (neural network, classical force field, or others). In a second step, these interaction potentials are employed in path integral molecular dynamics simulations, which is the formalism that incorporates the quantum nature of the nuclei in an efficient manner and allows to study the dynamic behavior of water in frameworks, its self-organization into various networks, and its effect on the mechanical stability of the framework. Model systems of interest are the commonly studied zirconium-based MOFs such as UiO-66(Zr) or MOF-801(Zr).
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsNanoporous materials, Nuclear quantum effects, machine learningReferences
[1] N. Hanikel, M.S. Prévot, F. Fathieh, E.A. Kapustin, H. Lyu, H. Wang, N.J. Diercks, T. Grant Glover, and O.M. Yaghi, ACS Central Science 5 (10), 1699-1706, 2019, 10.1021/acscentsci.9b00745
[2] Y. Sun, S.M.J. Rogge, A. Lamaire, S. Vandenbrande, J. Wieme, C.R. Siviour, V. Van Speybroeck, J.-C. Tan, Nat. Mater. 20, 1015–1023, 2021, 10.1038/s41563-021-00977-6
[3] N.C. Burtch, H. Jasuja, K.S. Walton, Chem. Rev. 114 (20), 10575-10612, 2014, 10.1021/cr5002589
[4] S. Batzner, A. Musaelian, L. Sun, M. Geiger, J.P. Mailoa, M. Kornbluth, N. Molinari, T.E. Smidt, B. Kozinsky, arXiv:2101.03164v3
Contact
Veronique Van Speybroeck