Developing a Hybrid Monte Carlo Algorithm for Peptides through Kernel Machine Learning
Developing a Hybrid Monte Carlo Algorithm for Peptides through Kernel Machine Learning
Promotor(en): T. Verstraelen, B. De Spiegeleer /25449 / Model and software developmentBackground and problem
Molecular dynamics (MD) simulations of biological macromolecules are used extensively in academia and commercial research centers to simulate small (nanometer-scale) fragments of living organisms. In such a simulation, the trajectory of each atom through time is determined by numerically solving Newton’s equation of motion using an approximate “force field” model to compute forces acting on the atoms. The macroscopic properties relevant for experimental research can, through the laws of statistical mechanics be extracted from the in-silico observations for these small model systems. In the past decades, especially the characterization of the different stable conformations of proteins or peptides (consisting of chains of amino acids) has been of great interest.
This thesis will focus on the NMG peptide, a (synthetic) opioid related to Spinorphin under active development in the group of Prof. B. De Spiegeleer. In the development of this compound, which is believed to be a good candidate for painkilling drugs, important questions remain open on the structure and activity of the compound, which could be solved through molecular modeling. Details on these peptides cannot be provided here due to intellectual property restrictions.
The main hurdle preventing these questions from being answered is that with the increasing size of molecules being simulated, the time scales of relevant processes such as transitions between different conformations becomes excessively long. Because MD simulations proceed with time steps of about 1 femtosecond, the simulation of a one-second process would take 1015 time steps, which is still infeasible today. The longest time scales which have been achieved in an MD simulation are of the order of milliseconds, using the ANTON computer located in the USA. [1]
However, instead of performing one long MD simulation, one may also characterize the dynamics of a macromolecule from many short MD simulations by making use of concepts from machine learning (kernels) and statistical physics (hybrid Monte Carlo methods and Markov state models).
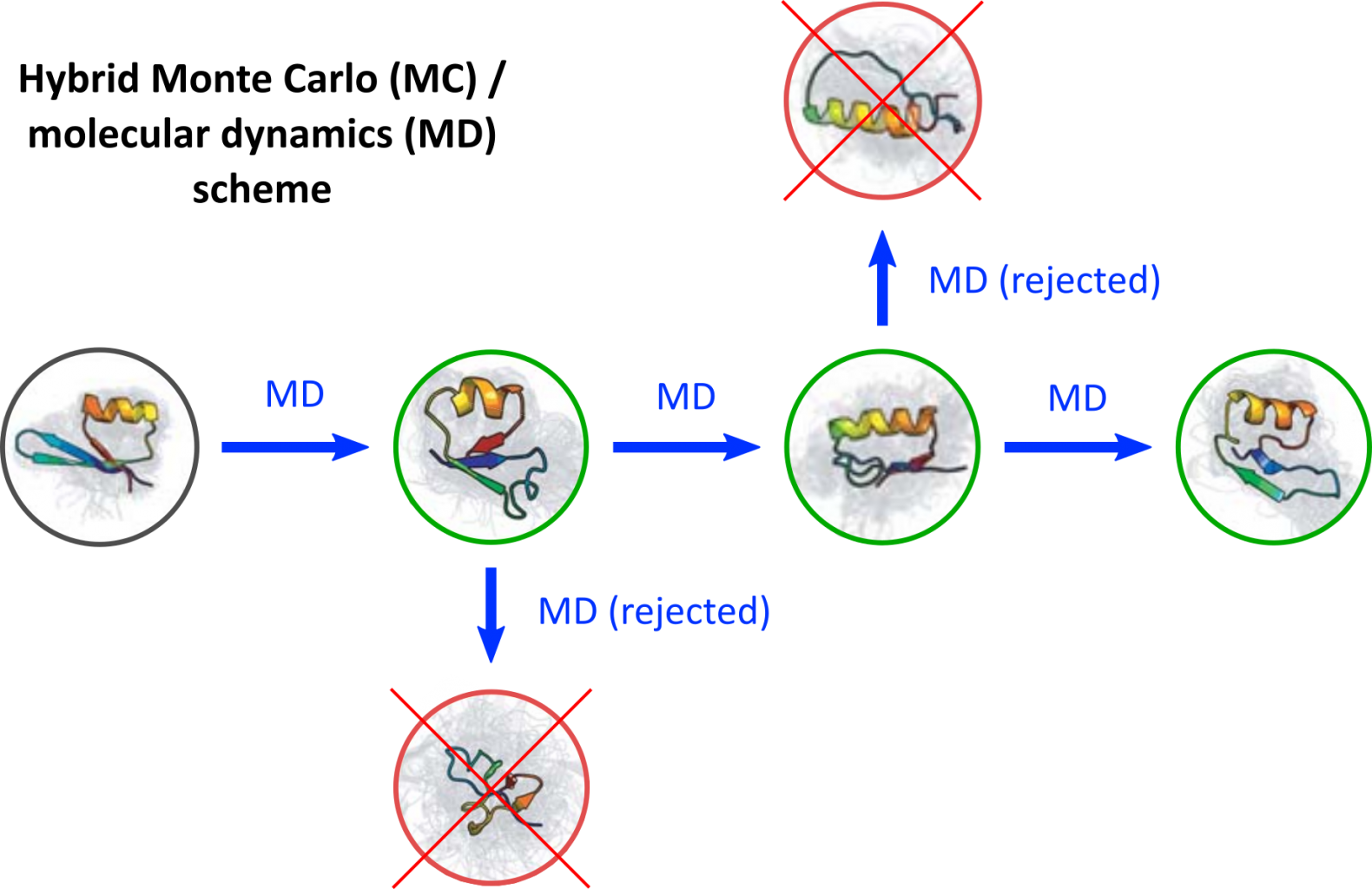
Goal
In order to distinguish between conformations of the peptide, a metric is required to represent the degree of similarity between two conformations. Usually, a root-mean-squared deviation is employed. A more advanced method is the use of a kernel, a technique from the world of machine learning. Specifically for use in peptides or proteins, such a kernel was proposed at the Center for Molecular Modeling which distinguishes conformations based on the set of dihedral angles present in the macromolecule.
To sample the full conformational space of the NMG peptide, a hybrid Monte Carlo approach will be followed in which short MD simulations (on the order of nanoseconds) represent the Monte Carlo moves, which are biased towards the transitions between stable conformations. This biasing can be achieved by using the previously mentioned kernel in kernel time-structure independent component analysis (KtICA), which is a technique to extract the slowest equilibrating modes from a simulation. [2] This will allow for a more efficient sampling of the transitions between conformations than performing a single long MD simulation.
Subsequently, using the MD simulations which were performed in the previous step, a Markov state model can be constructed. [3] This allows one to reduce the high-dimensional MD trajectories to a model containing a set of conformations and the transition probabilities (or timescales) between those conformations, answering most of the pertinent questions related to the structure and activity of the NMG peptide.
The main purpose of this thesis is to implement the kernel and use it in hybrid Monte Carlo simulations in order to efficiently simulate the system of interest, as well as to analyze the results using Markov state models (for which robust implementations are available in the literature).
List of figures: Hybride Monte Carlo (MC) / molecular dynamics (MD) scheme
- Study programmeMaster of Science in Biomedical Engineering [EMBIEN], Master of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsKernel Method, Hybrid Monte Carlo, Statistical physics, machine learning, PeptidesReferences
[1] D.E. Shaw et al., SC14 Proc., 43-53 (2014) https://dx.doi.org/10.1109/SC.2014.9
[2] C.R. Schwantes and V.S. Pande, J. Chem. Theory Comput., 11 (2015) https://doi.org/10.1021/ct5007357
[3] B.E. Husic and V.S. Pande, J. Am. Chem. Soc., 140, 7, 2386–2396 (2018)
https://doi.org/10.1021/jacs.7b12191
Contact
Toon Verstraelen