Derivation of coarse-grained models for large-scale simulations of flexible materials
Derivation of coarse-grained models for large-scale simulations of flexible materials
Promotor(en): T. Verstraelen, V. Van Speybroeck /MM_14_MODEV_07 / Model and software developmentNanoporous materials are a very attractive class of materials in modern science and industry. They are crystalline materials with nanosized pores that can accommodate for guest molecules. A very popular example of such materials are metal-organic frameworks (MOFs). MOFs are a hybrid class of materials in which inorganic metal oxides are interconnected by means of organic linkers. Except for this well-known type of material, new types of nanoporous materials are being developed every day, such as ZIFs, PoSiSils, … Such materials can be used for a variety of applications such as catalysis, gas storage, gas separation, gas detection, … Traditionally, MOFs are typically used as a powder. However, more and more scientists are interested in using these materials as a coating. Such coatings allow to add the typical functionalities of the nanoporous materials (adsorption, catalysis) to already existing devices, or they serve as a chemical protection for the underlying layers.
A very important property of these materials, especially when they are used as a coating, is their mechanical stability and flexibility. In other words, we are interested in answering the questions: “How does the material respond to pressure/stress?”, “How large is the induced strain?” and “Does the material collapse under pressure?”. To get accurate answers for these questions, we need to investigate these materials on a very high scale, i.e. thousands of unit cells. In this topic the student will try to answer these questions using molecular simulations. The most accurate simulations are atomic simulations in which the electrons are treated on a quantum mechanical level by solving the Schrödinger equation. However, such an approach is only feasible for small systems, typically less than 500 atoms. Since we want to simulate 1.000 unit cells and more, i.e. 100.000-1.000.000 atoms, we will need a different approach. A first huge upscaling is acquired by using so-called force fields, which do not treat electrons explicitly. Instead, the potential energy surface is approximated by means of empirical energy terms. These force fields allow to simulate systems of 1.000-10.000 atoms. A second upscaling is achieved by means of coarse-grained models. In such a model, not every atom is treated explicitly, instead we define beads which are interconnected and we postulate how the beads interact with each other, e.g. a harmonic spring between two neighboring beads (see figure). This is a very rough approximation of the molecular system, but it allows to describe the collective behavior of thousands of unit cells. The main goal of this thesis is to develop such coarse-grained models and use them to describe the mechanical behavior of nanoporous materials.
As a test case to develop the methodology for deriving coarse-grained models, we will study the metal-organic framework Mil-53. This is a flexible MOF that exhibits a very particular type of framework flexibility, i.e. breathing, in which the entire unit cell is able to expand and contract. This breathing effect can be triggered by means of several external stimuli such as temperature, pressure, adsorption, magnetic fields, ...Understanding this breathing phenomenon is very important to be able to control the behavior the MOF. In literature, there is already a lot of (mostly) experimental work available describing this breathing. However, a missing but crucial feature is the collective behavior of large single crystals. Coudert et al. reasoned that periodic boundary conditions used in MD simulations cannot describe the layer-by-layer transition between a large pore and a narrow pore phase of Mil-53 materials and that a multiscale approach is needed to capture the physics of breathing MOFs.
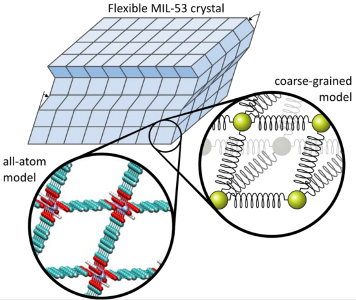
The Center for Molecular Modeling (CMM) has much experience in developing force fields for MOFs, especially for Mil-53, which resulted in the development of QuickFF: a methodology for deriving force fields from ab initio input. This methodology is implemented in a user-friendly Python package. The student will be able to use QuickFF for the derivation of the force fields in the first upscaling, and focus his/her attention on deriving a coarse-grained model from force-field simulations. For the derivation of a coarse-grained model, we will use a two-step procedure. In a first approximation, the module must reproduce the key features of the Mil-53 framework: e.g. the free energy difference between lp and np phases. In a second stage, the model will be refined with a larger training set of statistical observables obtained from atomistic force field simulations on Mil-53 super cells using NσijT Molecular Dynamics. These NσijT simulations are also being developed at the CMM and therefore, the student can rely on the experience present at the CMM. Note that in the second step, it is essential to use NσijT simulations to cover all the possible internal strains in the crystal. Once the coarse-grained model parameters are derived, a large isolated Mil-53 crystal model (thousands of unit cells) can be studied. On this sample, we can perform computational tensile strength tests and follow structural transitions up to length scales of 500 nm. Such simulations will offer a new perspective on several pressing issues related to flexible MOFs, e.g. “Can lp and np phases coexist in one Mil-53 crystal?”, or “Does this model confirm a layer-by-layer transition between lp and np phases?”.

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsCoarse graining, Force fields, Molecular simulation, Flexibility, Implementation, Analysis
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck
Toon Verstraelen