Computational investigation of the selective adsorption of gas mixtures in nanoporous materials
Computational investigation of the selective adsorption of gas mixtures in nanoporous materials
Promotor(en): L. Vanduyfhuys, V. Van Speybroeck /25469 / Model and software developmentBackground and problem
The separation of a gas mixture stream into its isolated components is a highly relevant process for many industrial applications such as refinement of crude oil and the production of high value chemicals and plastics. Many separation processes currently rely on cryogenic distillation in which differences in boiling points are exploited to separate constituents by selective condensation. Unfortunately, such distillation processes are very energy-demanding as they require either cooling down to very low temperatures (cryogenic distillation) or require the addition of the heat of vaporization. As a result, the current ecological and economical circumstances require more energy-efficient alternatives. A promising candidate is found in selective adsorption in nanoporous materials (illustrated in figure 1). Different gasses can have a different adsorption affinity (i.e. adsorption enthalpy) towards a given nanoporous materials, which implies that the component with the highest affinity would adsorb first and the component with the lowest affinity would remain in the gas stream. The application of such process is currently under investigation for the separation of propylene from propane or ethylene from ethane using membranes of nanoporous materials.
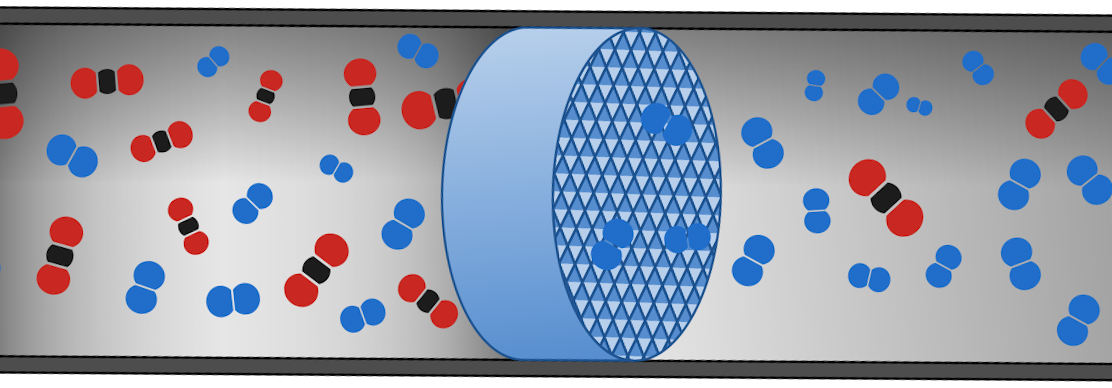
Figure 1: illustration of how a nanoporous material (blue cylinder in the middle) could separate CO2 (red-and-black molecules) from N2 (blue molecules) in an exhaust pipe.
One of the biggest advantages of using nanoporous materials is the huge number of candidate materials. In this topic, we will study metal organic frameworks (MOFs), which are hybrid materials consisting of inorganic metal-oxide bricks linked together through organic linkers (see figure 2). The resulting material represents a crystalline framework exhibiting empty voids or channels at the nanometer scale, hence nanoporous material. The building block structure allows for tailored design of new MOFs to fit the needs of a certain application. In the context of selective adsorption, we consider MOFs with an appropriate adsorption enthalpy for the relevant gasses. Unfortunately, the large number of possible MOFs also implies that an attempt to characterize them all using experimental techniques would be too expensive and time consuming. Therefore, the use of molecular simulation techniques to compute the properties and predict the behavior of these materials is indispensable [1]. Such computational characterization of the adsorption properties is the topic of this thesis subject.
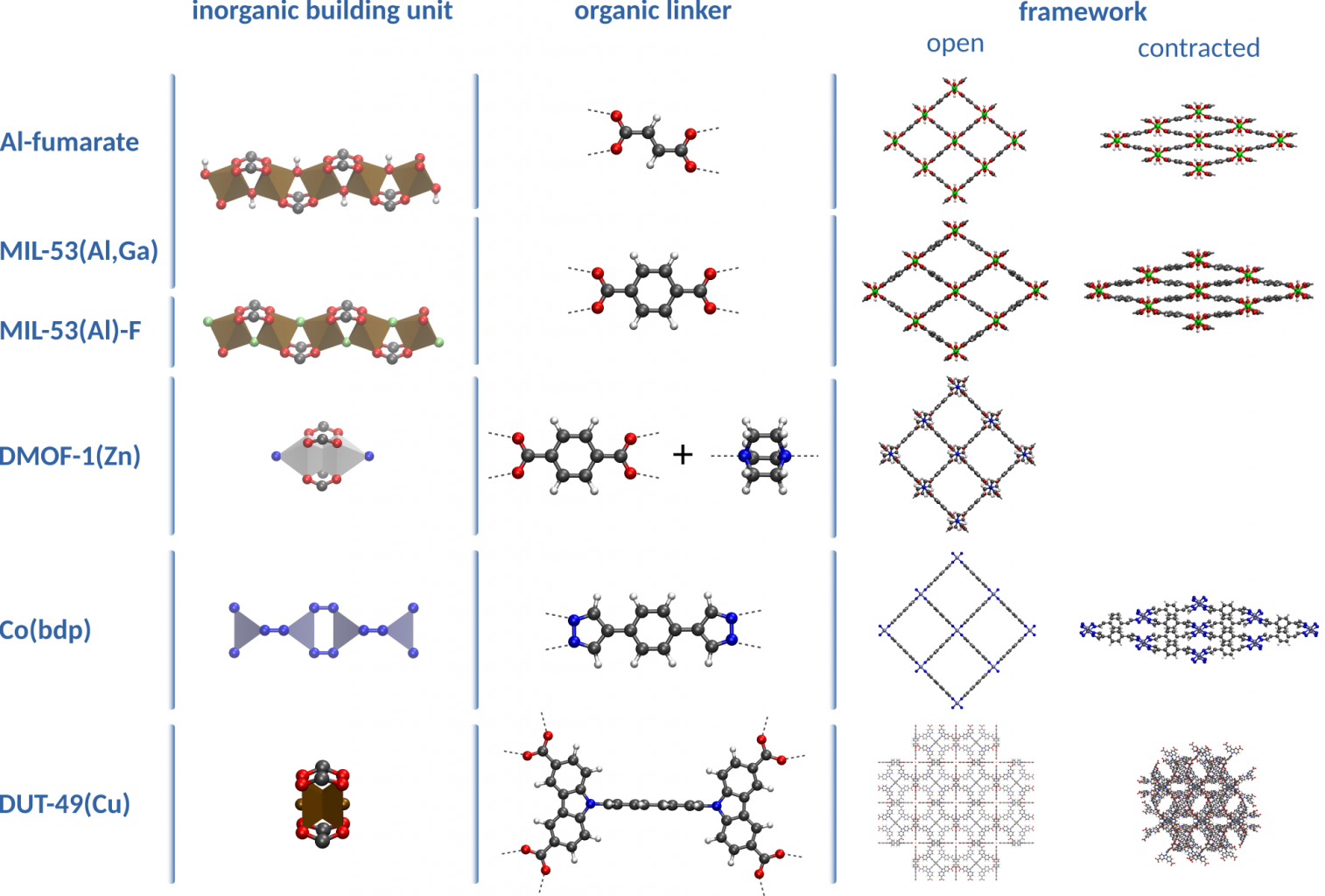
Figure 2: Illustration of the chemical structure of various metal-organic frameworks composing out of an inorganic building unit and an organic linker.
Goal
In this master thesis, the student will first get familiar with various molecular simulation techniques that allow to investigate adsorption properties. These techniques, such as grand canonical Monte Carlo (GCMC) [2] and classical Density Functional Theory (cDFT) [3], consider a periodic model for the MOF in thermodynamic equilibrium with a gas reservoir. Both methods then allow to compute the equilibrium density profile of adsorbed gas under the applied thermodynamic conditions (temperature and chemical potential). GCMC does this by subsequent steps of inserting/deleting gas molecules in the MOF until equilibrium is achieved. CDFT achieves the same thing by minimizing the grand potential (i.e. the thermodynamic potential in the grand canonical ensemble) as a functional of adsorbed density. Finally, by sweeping over the chemical potential (or equivalently the vapor pressure) at fixed temperature and computing the corresponding equilibrium adsorbed amount, one can construct the adsorption isotherm.
The student will apply these simulations to characterize the adsorption properties of pure gasses (e.g. pure nitrogen, ethane or ethylene) in various metal organic frameworks (such as ZIF-8). Next, he will investigate to what degree various adsorption theories such as ideal adsorption solution theory (IAST), osmotic framework adsorption solution theory (OFAST) [4] or an in-house developed thermodynamic model [5,6] can be applied to use the acquired pure-component adsorption isotherms and determine the adsorption selectivity of gas mixtures (i.e. the ratio of adsorbed amounts of the various components in the mixture). Depending on the interest of the student, the focus can shift more towards characterizing single-component adsorption properties of various molecules in various MOFs, or towards the application (and development) of adsorption theories for extracting the adsorption selectivity. This topic is directly related with the Moonrise project (https://moonshotflanders.be/mot3-moonrise/), an ongoing project in collaboration with various experimental groups in which energy-efficient alternatives for the cryogenic separation of e.g. ethane from ethene. As such, the student will have the opportunity to participate in scientific discussions and connect closely to the results of the ongoing experimental research at our experimental partners.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsAdsorption, isotherm, Statistical physics, Thermodynamics, classical density functional theory, grand canonical monte carloReferences
[1] L. Vanduyfhuys et al., Nature Commun., 2018, 9(1), 204
[2] S.M.J. Rogge et al., Adv. Theory Simulat., 2019, 2, 1800177
[3] Y. Liu and H. Liu, “Classical Density Functional Theory for Fluids Adsorption in MOFs”, chapter in “Metal Organic Frameworks”, F. Zafar & E. Sharmin, IntexOpen, 2016
[4] F. Coudert, Phys. Chem. Chem. Phys., 2010, 12(36), 10904
[5] L. Vanduyfhuys et al., Mol. Simulat., 2015, 41(16), 1311
[6] L. Vanduyfhuys and G. Maurin, Adv. Theory Simulat., 2019, 2, 1900124
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck