Computational CO2 capture from first principles
Computational CO2 capture from first principles
Promotor(en): T. Verstraelen, V. Van Speybroeck /MM_14_MODEV_04 / Model and software development, Nanoporous materialsThe main reason for the current climate change is that our economy is based on non-sustainable technologies. The excessive emission of carbon dioxide in the environment is an unavoidable consequence of the quality of life we are used to. For this reason, there is a large interest in new carbon (CO2) capture technologies that can be installed at the emission source to prevent an uncontrolled release in the atmosphere.
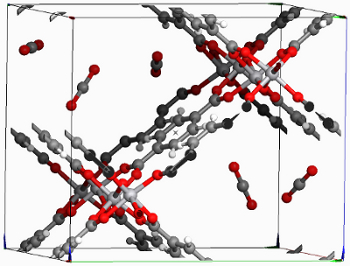
Figure: CO2 molecules in the MIL-47 framework One possible solution is the selective adsorption of CO2 in crystalline porous materials like zeolites or metal-organic frameworks, followed by extraction and storage. Due to the large variety of these materials and the uncountable number of hypothetical structures, it is no longer feasible to synthesize and test each material in a lab experiment until a promising candidate is found. Instead, one can test the CO2 adsorption in computer simulations at a much faster rate. The models used to characterize the interactions between the inner surface of the porous material and a CO2 molecule still rely on experiments. Especially for simulations on hypothetical materials, it is desirable to obtain the interaction parameters directly from quantum mechanical electronic structure models.
Several non-empirical models to describe the interactions between guest molecules and a porous framework are currently investigated at the Center for Molecular Modeling (CMM). The models and their parameters are solely based (directly and indirectly) on the Schrödinger equation and some fundamental constants. Starting from a DFT computation on the empty host framework, parameterized models for electrostatic, London dispersion and Pauli repulsion interactions are derived. These models will be tested for the prediction of the adsorption isotherms of CO2 and N2 with grand-canonical Monte Carlo simulations. The final aim of this thesis is to select the most appropriate techniques for a massive screening of porous materials. The simulation software needed for this project will be provided.

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsModel development, Non-bonding interactions, Carbon capture, Monte CarloRecommended coursesComputational physics (C001827); Modeling and simulation at the nanoscale (E023370)
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck
Dr. ir. Lennart Joos
Toon Verstraelen