Chemische kinetiek van basisch gekatalyseerde reacties in metaal-organische roosters
Chemische kinetiek van basisch gekatalyseerde reacties in metaal-organische roosters
Promotor(en): V. Van Speybroeck, K. Hemelsoet /MM_14_NANO_02 / Nanoporous materials(Important remark: the thesis can be written in Dutch or English, as the student prefers)
Metal-Organic Frameworks (MOFs) are - just as zeolites - ordered nanoporous materials with pore sizes between 0.5 and 2 nm. In contrast to zeolites, which have a purely inorganic character, MOFs constitute a new class of porous materials that exhibit a truly hybrid character, since both inorganic and organic moieties are needed to build up their framework. While catalytic conversions and separations are the most frequently used industrial applications of zeolites, future MOF applications lie more in adsorption, storage and separation of gas mixtures (H2, CO2 or CH4), and nowadays their importance in catalysis is steadily increasing. Although catalysis is potentially one of the most important applications of these exciting class of materials, design of suitable MOFs for catalytic applications remains a challenge to be explored.
This thesis subject is focused on reactions in basic-MOFs, such as M2(BTC)(NO3) (M= Sr and Ba, BTC= Benzene Tri Carboxylate, Figure 1) and is in fact a collaboration with the Centre for Surface Chemistry and Catalysis, Prof. D. De Vos - KULeuven. During an activation treatment at high temperatures and elevated pressure, active sites are created.
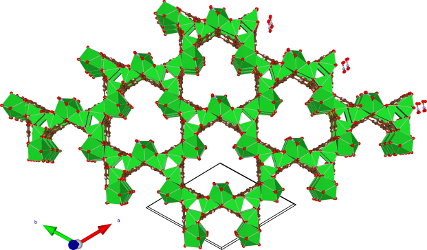
Figure 1.Initial experiments on the Ba and Sr-MOFs have already shown the modulating effect of the metal on the conversion of the Knoevenagel condensation of malonitrile and benzaldehyde (reaction 1, Figure 2). However, the exact mechanism of the base catalyzed reactions still needs to be determined.
Within this thesis, attention will be given to basic MOFs and their use as catalysts. Therefore, the nature of the basic sites will have to be unraveled by spectroscopic probe molecules and chemical reactions. To characterize the acidity and basicity of the active sites, we can use probe molecules such as CO for acid sites and pyrrole for base sites. These can offer a first indication about the strength of the active sites and the possible adsorption modes. As a result frequency shifts in the IR-spectra are obtained. To properly model these properties, periodic Density Functional Theory calculations in which the full molecular environment is taken into account, are best suited.
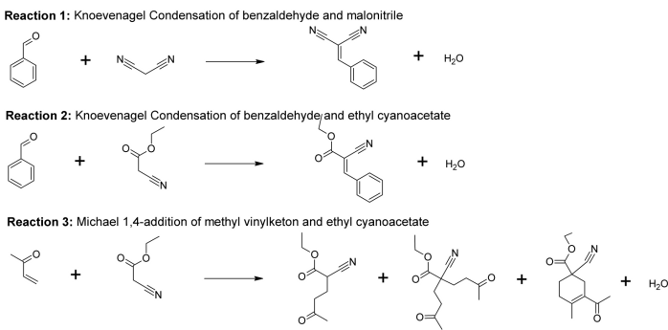
Figure 2.Experimental data of three reactions (shown in Figure 2) is already available (from the Center for Surface Chemistry and Catalysis, KULeuven). Molecular modeling techniques will be used to unravel the mechanism of the base catalyzed reactions on well-chosen molecular models (cluster and periodic models). As a first step - the deprotonation of one reactant at a base site (Reactions 1, 2, 3) might be required, forming a carbanion adsorbed on an adjacent metal site. Reaction 1 should go faster than reaction 2, based on the pKa of the nitriles (pKa(malonitrile)=11, pKa(ethyl cyanoacetate)=9) that deprotonate before reaction. A Michael 1,4-addition (reaction 3, Figure 2) will also be studied as this type of reaction belongs to the larger class of conjugate additions and is one of the most useful strategies for the mild formation of C-C bonds. This reaction can be seen as a nucleophilic addition of a carbanion of ethyl cyanoacetate with methyl vinylketon.
Summarizing, a modeling study should be performed to complement existing experimental data and to provide insight into the nature of the active site. A variety of theoretical techniques (encompassing cluster and periodic calculations) are available at the Center for Molecular Modeling (CMM).


- Study programmeMaster of Science in Chemical Engineering [EMCHEM]
Contact
Prof. Dr. ir. Karen Hemelsoet
Veronique Van Speybroeck
Matthias Vandichel