Mechanistic studies of aldol condensations in UiO-66 and UiO-66-NH2 metal organic frameworks
Abstract
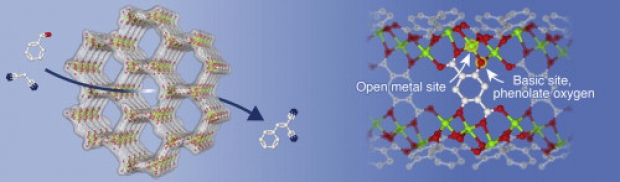
A full mechanistic investigation is proposed for the industrially important cross-aldol condensation reaction of heptanal with benzaldehyde on the UiO-66 and the amino-functionalized UiO-66-NH2 metal–organic frameworks to form jasminaldehyde. Several experimental studies indicate that the activity for the aldol condensation reaction can be increased by proper functionalization of the material, e.g. by introducing an additional basic amino site and thus creating a bifunctional acid–base catalyst for the aldol condensation. The precise molecular level origin for this behavior is to date unclear. Herein state-of-the-art Density-Functional Theory (DFT) calculations have been performed to unravel the mechanism of the cross- and self-aldol condensations of benzaldehyde and propanal. To this end free energy calculations have been performed on both extended cluster and periodic models. It is found that the mechanism on both catalysts is essentially the same, although a slightly stronger adsorption of the reactants and slightly lower barriers were found on the amino functionalized material, pointing toward higher initial activities. New experiments were performed to confirm these observations. It is indeed found that the initial activity toward cross-aldol condensation on the amino functionalized material is higher, although after about 40 min of reaction both materials become equally active. Our results furthermore point out that the basic amino groups may promote side reactions such as imine formation, which is induced by water. The study as presented can assist to engineer materials at the molecular level toward the desired products.
 Open Access version available at UGent repository
Open Access version available at UGent repository