Mechanistic characterization of zeolite-catalyzed aromatic electrophilic substitution at realistic operating conditions
Abstract
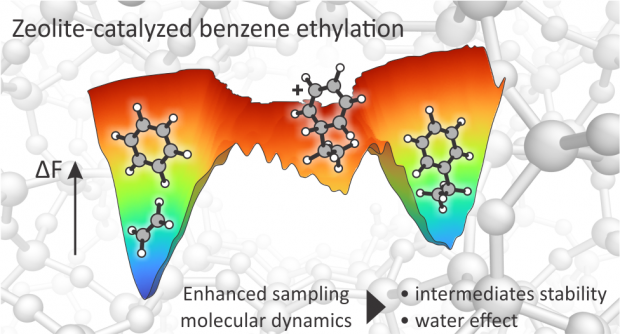
Zeolite-catalyzed benzene ethylation is an important industrial reaction, as it is the first step in the production of styrene for polymer manufacturing. Furthermore, it is a prototypical example of aromatic electrophilic substitution, a key reaction in the synthesis of many bulk and fine chemicals. Despite extensive research, the reaction mechanism and the nature of elusive intermediates at realistic operating conditions is not properly understood. More in detail, the existence of the elusive arenium ion (better known as Wheland complex) formed upon electrophilic attack on the aromatic ring is still a matter of debate. Temperature effects and the presence of protic guest molecules such as water are expected to impact the reaction mechanism and lifetime of the reaction intermediates. Herein, we used enhanced sampling ab initio molecular dynamics simulations to investigate the complete mechanism of benzene ethylation with ethene and ethanol in the H-ZSM-5 zeolite. We show that both the stepwise and concerted mechanisms are active at reaction conditions and that the Wheland intermediate spontaneously appears as a shallow minimum in the free energy surface after the electrophilic attack on the benzene ring. Addition of water enhances the protonation kinetics by about 1 order of magnitude at coverages of one water molecule per Brønsted acidic site. In the fully solvated regime, an overstabilization of the BAS as hydronium ion occurs and the rate enhancement disappears. The obtained results give critical atomistic insights in the role of water to selectively tune the kinetics of protonation reactions in zeolites.

