Cation−π Interactions Accelerate the Living Cationic Ring-Opening Polymerization of Unsaturated 2-Alkyl-2-oxazolines
Abstract
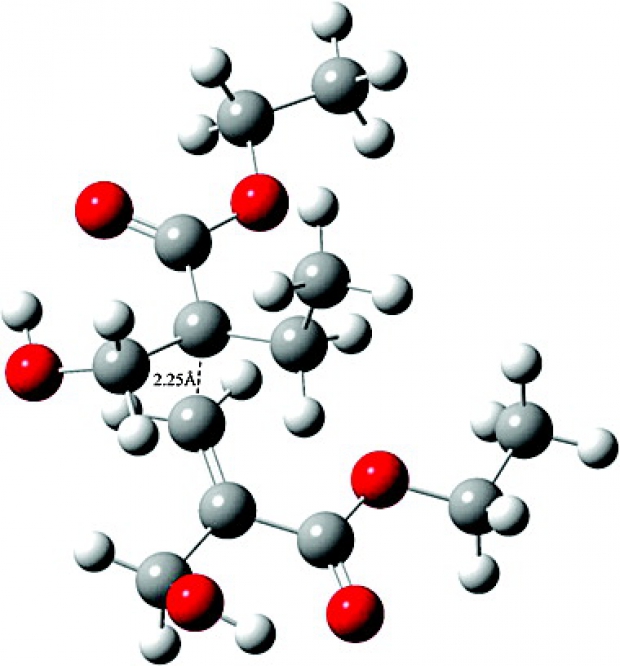
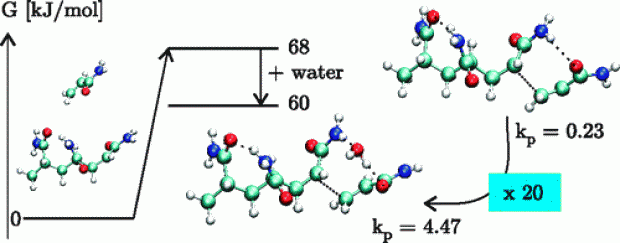
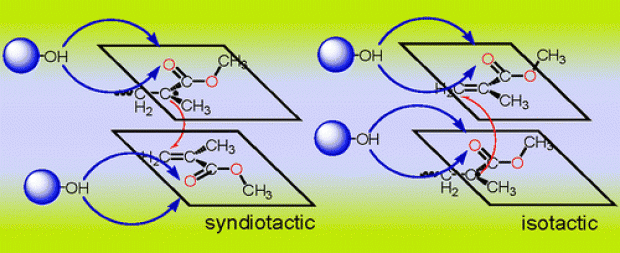
Cation–dipole interactions were previously shown to have a rate-enhancing effect on the cationic ring-opening polymerization (CROP) of 2-oxazolines bearing a side-chain ester functionality. In line with this, a similar rate enhancement—via intermolecular cation−π interactions—was anticipated to occur when π-bonds are introduced into the 2-oxazoline side-chains. Moreover, the incorporation of π-bonds allows for facile postfunctionalization of the resulting poly(2-oxazoline)s with double and triple bonds in the side-chains via various click reactions. Herein, a combined molecular modeling and experimental approach was used to study the CROP reaction rates of 2-oxazolines with side-chains having varying degrees of unsaturation and side-chain length. The presence of cation−π interactions and the influence of the degree of unsaturation were initially confirmed by means of regular molecular dynamics simulations on pentameric systems. Furthermore, a combination of enhanced molecular dynamics simulations, static calculations, and a thorough analysis of the noncovalent interactions was performed to unravel to what extent cation−π interactions alter the reaction kinetics. Additionally, the observed trends were confirmed also in the presence of acetonitrile as solvent, in which experimentally the polymerization is performed. Most intriguingly, we found only a limited effect on the intrinsic reaction kinetics of the CROP and a preorganization effect in the reactive complex region. The latter effect was established by the unsaturated side-chains and the cationic center through a complex interplay between cation−π, π–π, π–induced dipole, and cation–dipole interactions. These findings led us to propose a two-step mechanism comprised of an equilibration step and a CROP reaction step. The influence of the degree of unsaturation, through a preorganization effect, on the equilibration step was determined with the following trend for the polymerization rates: n-ButylOx < ButenOx < ButynOx ≥ PentynOx. The trend was experimentally confirmed by determining the polymerization rate constants.
 Open Access version available at UGent repository
Open Access version available at UGent repository