Minimal Basis Iterative Stockholder: Atoms-in-Molecules for Force-Field Development
Abstract
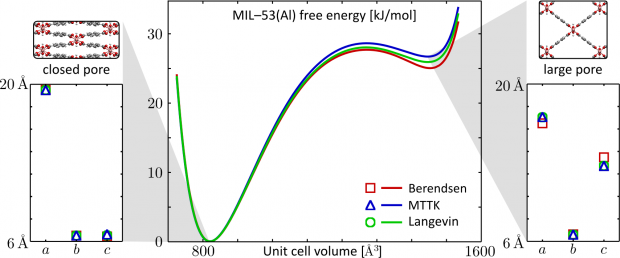
Atomic partial charges appear in the Coulomb term of many force-field models and can be derived from electronic structure calculations with a myriad of atoms-in-molecules (AIM) methods. More advanced models have also been proposed, using the distributed nature of the electron cloud and atomic multipoles. In this work, an electrostatic force field is defined through a concise approximation of the electron density, for which the Coulomb interaction is trivially evaluated. This approximate "pro-density" is expanded in a minimal basis of atom-centered s-type Slater density functions, whose parameters are optimized by minimizing the Kullback-Leibler divergence of the pro-density from a reference electron density, e.g. obtained from an electronic structure calculation. The proposed method, Minimal Basis Iterative Stockholder (MBIS), is a variant of the Hirshfeld AIM method but it can also be used as a density-fitting technique. An iterative algorithm to refine the pro-density is easily implemented with a linear-scaling computational cost, enabling applications to supramolecular systems. The benefits of the MBIS method are demonstrated with systematic applications to molecular databases and extended models of condensed phases. A comparison to 14 other AIM methods shows its effectiveness when modeling electrostatic interactions. MBIS is also suitable for rescaling atomic polarizabilities in the Tkatchenko-Sheffler scheme for dispersion interactions.