Semi-analytical thermodynamic model for multicomponent adsorption in nanoporous materials
Semi-analytical thermodynamic model for multicomponent adsorption in nanoporous materials
Promotor(en): V. Van Speybroeck, A. Ghysels /17NANO13 / Nanoporous materialsNanoporous materials are a very promising class of materials, which attract a lot of attention from both academia and industry. They are crystalline materials that contain pores with a size of several nanometers. In many types of their applications, it is crucial that guest molecules can adsorb to the inner surface due to favorable host–guest interactions. This gives rise to applications such as storage, detection and separation of gases as well as catalysis of chemical reactions. Two classes of such nanoporous materials are zeolites, i.e. inorganic frameworks consisting of Si, O and Al, and Metal-Organic Frameworks (MOFs), i.e. hybrid frameworks consisting of inorganic metal-oxide clusters interconnected by means of organic linkers. For example, zeolites are used in the energy sector to harvest waste heat and MOFs have been successfully used to store hydrogen much more efficiently and safely than pressurized cans. Moreover, in the case of MOFs, adsorption of guest molecules can induce drastic variations of the size and shape of the unit cell. This phenomenon is called breathing; a MOF can sequentially shrink and expand again when increasing the loading of guest molecules.
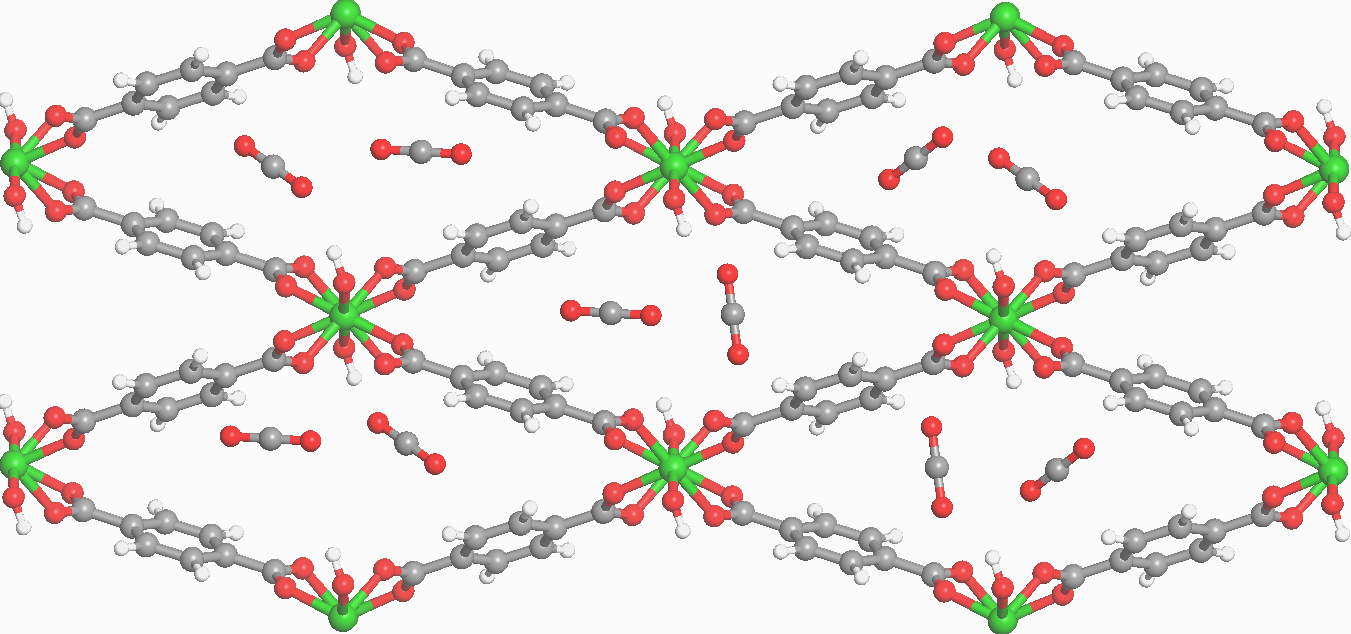
Due to the immense amount of various nanoporous materials, an experimental characterization of all materials is impossible. Therefore, a computational screening of existing and hypothetical materials is very useful to identify promising candidates for certain applications. The most accurate calculations involve a quantum mechanical description of both the host framework as well as the guest molecules. However, such an approach usually requires an a priori knowledge of the loading, i.e. the amount of guest molecules that adsorb in the pores. The situation becomes even more complicated when mixtures of gases are responsible of the loading as not only the total loading but also the composition of the guest molecules in the pores is relevant. In most computational studies up to now, the loading is limited to a single molecule or one estimates the number of guest molecules from experiment. However, in the spirit of accurate screening of hypothetical materials, such an approach is no longer sufficient.
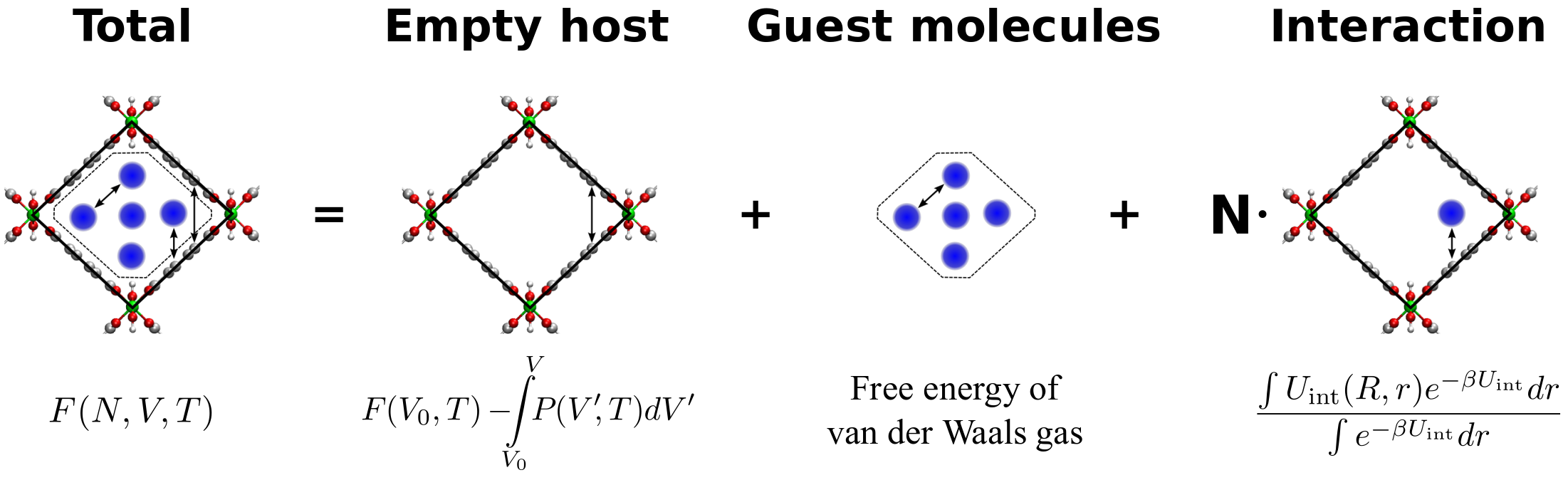
A new thermodynamic model was recently developed at the Center for Molecular Modeling that accurately predicts the loading of single component adsorption in nanoporous materials. This model starts from the output of molecular dynamics (MD) and Monte Carlo (MC) simulations, describing the free energy as a function of the fixed unit cell volume, number of guest molecules and temperature. By applying the Legendre transformation, this free energy (canonical ensemble) can be transformed to the so-called osmotic potential in terms of chemical potential and the mechanical pressure. As such, the number of adsorbed guest molecules as well as the unit cell size and shape can be computed as a function of the chemical potential and mechanical pressure. As mentioned before, this model is currently only suited for describing single-component adsorption and needs to be extended to multicomponent adsorption.
Goal
In this work the student will extend the thermodynamic model to account for adsorption of multiple species. The free energy needed as input for the model contains a term describing the interaction between guest molecules. This term needs to be extended to account for multiple species by introducing the interaction between the species. Several approaches can be attempted, ranging from more theoretical derivations that start from the van der Waals expression for a single-component gas, to more computationally inspired approaches were we perform multiple molecular simulations of single- and multi-component gases and try to find a general analytic model. Furthermore, extra Legendre transformations will be required to account for the chemical potential (or vapor pressure) of each adsorbing component.
This thesis is a mixture of macroscopic thermodynamics, statistical physics, mathematical modeling and molecular simulation. The student will need to study the problem, come up with a suitable model, implement it in basic scripts and test it extensively. Programming skills will be transferred during the thesis if it is required.
Aspects
Physics: this subject requires a fundamental knowledge of thermodynamics and statistical physics to be able to capture all important features of the interactions in the thermodynamic model.
Engineering: applying the resulting thermodynamic model allows to characterize the adsorption properties of nanoporous materials in an efficient and accurate way.


- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELLING, NANOKeywordsThermodynamics, Statistical physics, van der Waals gas, Adsorption, Free energy calculations
Contact
Louis Vanduyfhuys
An Ghysels
Veronique Van Speybroeck