Obtaining the thermal conductivity of metal-organic frameworks via force-field molecular dynamics simulations
Obtaining the thermal conductivity of metal-organic frameworks via force-field molecular dynamics simulations
Promotor(en): V. Van Speybroeck /17NANO09 / Nanoporous materialsMetal-organic frameworks (MOFs) [1] are a unique class of porous crystalline materials, which are made up by inorganic moieties connected by organic linkers (see Figure 1). The materials can be synthesized with a large versatility and can easily be tuned, both chemically and physically, for specific applications. This new class of materials has received enormous attention from academia and industry as they show an exceptional potential for energy storage, catalysis, energy-related gas separations, controlled drug release, nano-shock absorbers and -dampers, …
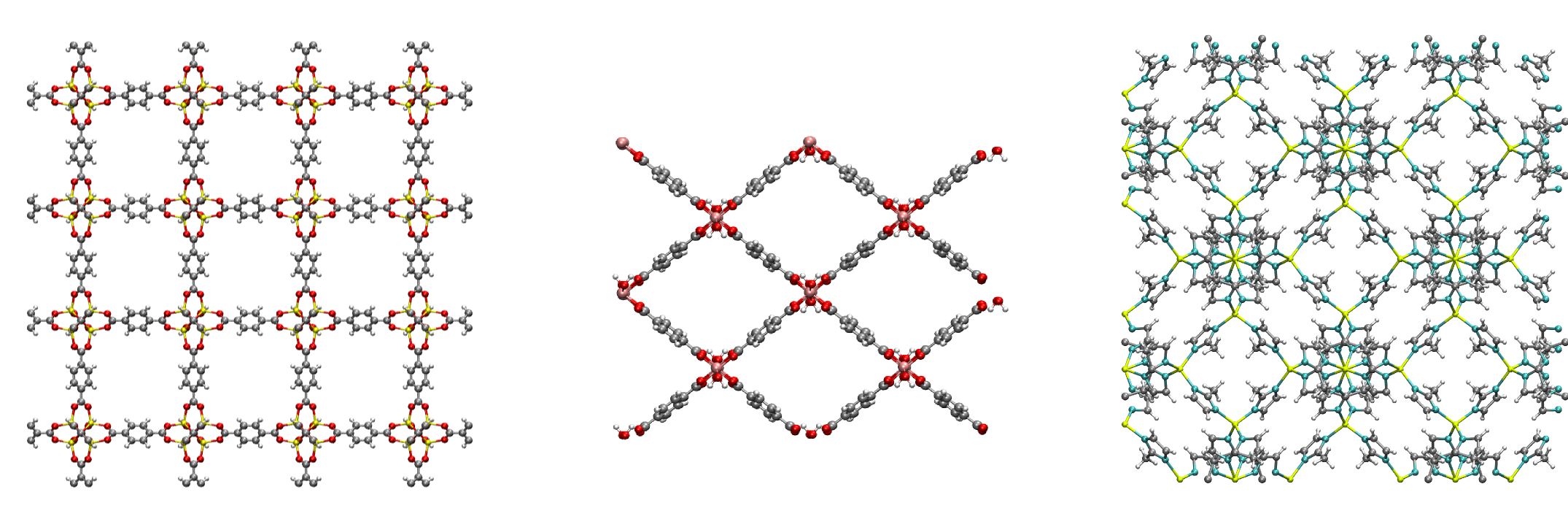
Figure 1. View along the -direction of MOF-5 (left), MIL-53 (middle) and ZIF-8 (right)However, a thermal characterization of MOFs is not yet available, although for many applications knowledge on properties such as the heat capacity and thermal conductivity is crucial. This proposal aims to fill this gap by focusing on the development and application of a toolbox to study thermal transport properties.
Thermal conductivity relates the heat current J to the temperature gradient ∇T via the macroscopic Fourier law J = κ∙∇T in which κ is the thermal conductivity tensor. It is experimentally very hard to extract information from this relation in a laboratory setting. Using atomistic simulations, we will try to obtain this tensor. These simulations are computationally too expensive when electronic structure methods are used such as Hartree-Fock or density functional theory. Alternatively, one can use so-called force fields [2], in which the complex potential energy surface is approximated by simple analytical expressions inspired by simple spring models.Goal
In this thesis, the student will perform molecular simulations using force fields to compute the thermal conductivity tensor of several metal-organic frameworks. An approach to model the thermal conductivity is through equilibrium molecular dynamics simulations. The temperature is a macroscopic property related to the thermal motion of the atoms. During a molecular dynamics simulation at a fixed temperature, energy, and thus also heat, is exchanged between the atoms of the material, while integrating the Newtonian equation of motion. While it might seem counterintuitive to calculate the thermal conductivity, which is a transport property, from a simulation mimicking thermodynamic equilibrium, it is possible to do so through the application of the Green-Kubo theorem:
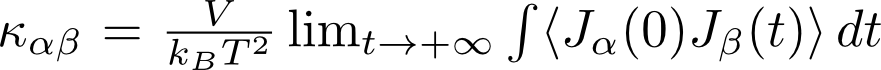
Here, non-equilibrium transport properties are related to microscopic equilibrium fluctuations of the system and thus without strictly applying a macroscopic temperature gradient, information is extracted on the thermal conductivity.
In literature, this has been done for various simple systems such as carbon and silica materials, but only seldom for MOFs. At first, we will try to reproduce the available theoretical and experimental results of MOF-5 and ZIF-8 (e.g. [3]) to validate the methodology. In the next step, it will be applied to highly relevant MOFs such as the rigid UiO-66 and the flexible MIL-53 and MIL-47. Especially these last MOFs can lead to interesting new insights, as they display a large structural flexibility under various stimuli.
The Center for Molecular Modeling has ample expertise in modeling nanoporous materials and metal-organic frameworks and performs this research in the framework of large scale European and international research programs. For the emphasized materials we developed various force fields and predicted a multitude of physical and chemical properties with success. The modeling is performed in close collaboration with experimental partners to achieve an optimal synergy between modeling and experimental efforts. The student will be actively involved in the work discussions with these international prominent partners. Furthermore the student will be actively coached to get acquainted as soon as possible with all necessary techniques to perform the suggested research successfully.
Context Engineering
Physics: explore the quantum-mechanical potential energy surface via molecular dynamics simulations and apply statistical physics to obtain the macroscopic thermal conductivity.
Engineering: development of a methodology to model the thermal conductivity.

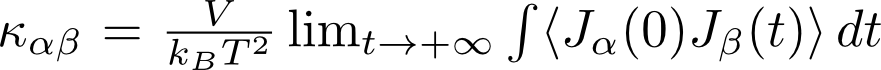
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELLING, NANOKeywordsThermodynamics, Statistical physics, thermal transport, Molecular simulation, Nanoporous materialsReferences
[1] H.-C. Zhou, S. Kitagawa, Metal-organic frameworks (MOFs), Chem. Soc. Rev. 2014, 43, 5415-5418
[2] L. Vandufhuys, S. Vandenbrande, T. Verstraelen, R. Schmid, M. Waroquier, V. Van Speybroeck, QuickFF: a program for a quick and easy derivation of force fields for metal-organic frameworks from ab initio input, J. Comput. Chem. 2015, 36, 1015-1027
[3] X. L. Zhang, J. W. Jiang, Thermal conductivity of zeolitic imidazolate framework-8: a molecular simulation study, J. Phys. Chem. C 2013, 117, 18441-18447
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck