Designing 2D hybrid organic-inorganic perovskites for game-changing photovoltaics
Designing 2D hybrid organic-inorganic perovskites for game-changing photovoltaics
Promotor(en): V. Van Speybroeck, K. Lejaeghere /17SPEC02 / SpectroscopyPerovskites are well-known minerals with a ABX3 stoichiometry. Named after Russian mineralogist Lev Perovski, their orthorhombic crystal structure has already been identified in the early 20th century. Because of their strong coupling between crystal symmetry and directional materials properties (e.g. dipole moment), perovskites have attracted renewed interest: they exhibit exciting new properties such as ferro-electricity (spontaneous polarization) and ferro-elasticity (spontaneous strain).
One particular class of perovskites, hybrid organic-inorganic perovskites (HOIPs), are now under world-wide investigation for an entirely different reason: they can be applied for photovoltaic (PV) purposes. HOIPs contain both inorganic and organic components. The most famous HOIP currently used is methylammonium (MA) lead iodide (MAPbI3), where the organic methylammonium cation is encaged in a negatively charged lead iodide framework. Since its first consideration as a PV material in 2009, the efficiency of perovskite-based solar cells has gone up from 3.8 % to 22.1 %. This strong growth over only a few years makes many researchers believe that there is still room for improvement. Their easy synthesis, low cost and tunable band gap make them promising competitors for traditional PV materials.
HOIPs have some disadvantages as well, however. The most important concern is the low resistance of HOIPs to moisture. Water easily penetrates the lead iodide cages and breaks down the perovskite crystal structure. A possible solution was discovered only 3 years ago. Using so-called Ruddlesden-Popper phases allows a drastic limitation of the sensitivity of HOIPs to water. These special forms of perovskites consist of 2D layers of traditional perovskite material, interleaved with cations. In the case of 2D HOIPs, these spacer cations are organic as well, with the most prominent example being MAPbI3 interleaved with n-butylammonium (BA) (see figure). The organic tails coming out of the perovskite layers effectively keep out water, explaining the increased resistance to moisture. Thus far, the best available 2D HOIP produced has an efficiency of 12.52 %, but tuning the used organic and inorganic components of the structure should make it possible to considerably increase this efficiency further.
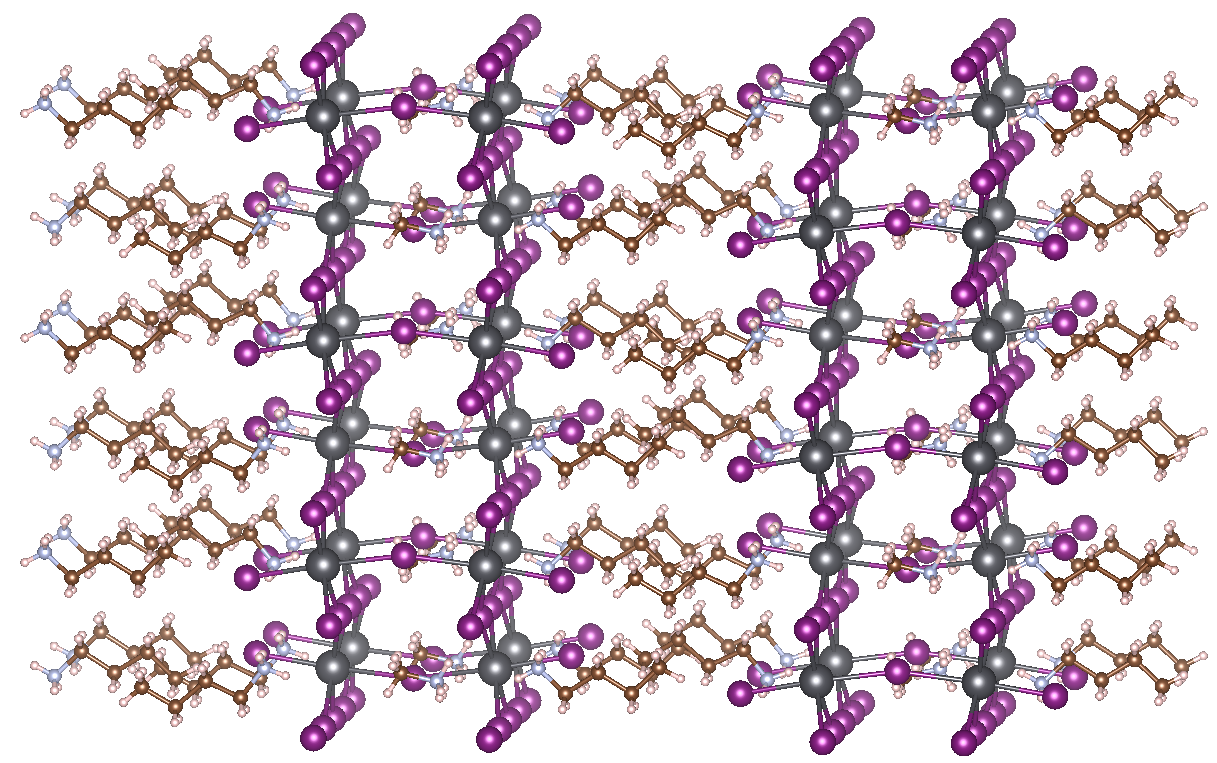
Goal
The goal of this thesis is to characterize and design 2D HOIPs using quantum physical software, using density-functional theory (DFT), such as the VASP package. Your objective is to understand the interactions which govern the geometry and band structure of these materials, in order to propose PV crystals with a potential improved efficiency.
When considering the current state of the art in 2D HOIPs, the best way to improve the efficiency of these PV materials even further, is to modify the spacer cations that separate the perovskite layers. However, it is not clear how strongly the spacer cations may be changed before the perovskite template is not able to support them anymore. In regular 3D HOIPs, there are tolerance factors, which describe the stability of the perovskite framework in terms of the size of the encaged organic cation. An important first objective of your thesis is to investigate whether a similar rule of thumb can be established for 2D HOIPs. To do this, you will start from the best known 2D HOIP, (BA)2(MA)3Pb3I10, and increasingly add to the width of the butylammonium ion by adding functional groups to the sides of the butyl chain. The stability of the perovskite framework can then be assessed in terms of its formation energy and deformation modes.
A second factor that influences the efficiency of solar-cell materials is the electronic structure. The band gap must be tuned to the energy of visible light, and the energy levels of the different components of the structure must be aligned in such a way that they allow efficient charge transfer. Your second objective is to derive trends that describe the relation between the used spacer cations and the resulting electronic structure. It has been shown in literature that the description of the band structure of lead iodide perovskites requires the inclusion of self-energy corrections and spin-orbit coupling. You will need to perform some tests to see to what extent these contributions can be determined from calculations on simplified crystals and whether they can be considered as independent effects (which can hence be computed separately).
Finally, an important aspect to keep in mind is that realistic materials contain defects. The 3D MAPbI3, for example, is known to have iodide vacancies (missing iodide) and interstitial lead atoms (additional lead in between lattice positions). Missing spacer cations are a type of defect that can only be found in 2D layered perovskites, and which have therefore not been investigated yet. If time allows, you will study how the presence of defects influences the geometry, stability and electronic structure of 2D HOIPs. Defects may lead to unfavourable effects, but can also be used to tune the electronic properties of materials, similar to doping in well-known semiconductors such as silicon or gallium arsenide.
Context for Engineering Physics students
Physics: use of quantum mechanical models for materials modelling
Engineering: application to materials properties and semiconductor engineering

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELLING, PHOTONICS, MATERIALS, NANO, ELECTRONICSKeywordshybrid inorganic-organic perovskite, density-functional theory, stability, electronic structure, defectsReferences
W.-J. Yin et al., ‘Halide perovskite materials for solar cells: a theoretical review’, J. Mater. Chem. A 3, 8926-8942 (2015). http://dx.doi.org/10.1039/C4TA05033A.
L. Pedesseau et al., ‘Advances and Promises of Layered Halide Hybrid Perovskite Semiconductors’, ACS Nano 10, 9776-9786 (2016). http://dx.doi.org/10.1021/acsnano.6b05944.
Contact
Veronique Van Speybroeck