Computational and experimental characterization of the structural and vibrational degrees of freedom of covalent organic frameworks
Computational and experimental characterization of the structural and vibrational degrees of freedom of covalent organic frameworks
Promotor(en): V. Van Speybroeck, H. Vrielinck /17SPEC01 / SpectroscopyIn recent years, crystalline nanoporous materials such as metal-organic frameworks (MOFs) and covalent organic frameworks (COFs) have been widely investigated for their potential use in various applications such as gas storage, gas separation, catalysis of chemical reactions and drug delivery systems. The main features of these materials allowing for such applications are their large internal pores and long-range ordered structure, allowing them to host various guest molecules adsorbed inside these pores. In contrast to MOFs, which have already been investigated for almost three decades, COFs are a much more recent subject of scientific research. In 2005, it was first shown that it is possible to assemble organic building units towards crystalline porous materials, which marks the birth of covalent organic frameworks [1]. COFs may be fabricated from planar 2D sheets or from non-planar building units, yielding 2D or 3D COFs respectively. Due to the absence of metals, COFs are much lighter than many other porous materials such as MOFs, while they still possess very high surface areas and are chemically and thermally very stable. Although the absence of metals also limits their use as catalysts, the large internal pores allow them to adsorb large activated molecules and fix them on the framework, a process called anchoring, which in turns opens up new pathways for including new functionalities to the materials. However, an open question remains if the original materials are mechanically stable/flexible enough to include/tolerate such large molecules. In the case of COFs that consist of stacked 2D sheets, for example, the ease with which adjacent sheets can shear over each other is expected to influence the shape and size of the possible channels/pores, which in turn will influence the penetration depth of the anchor molecules.
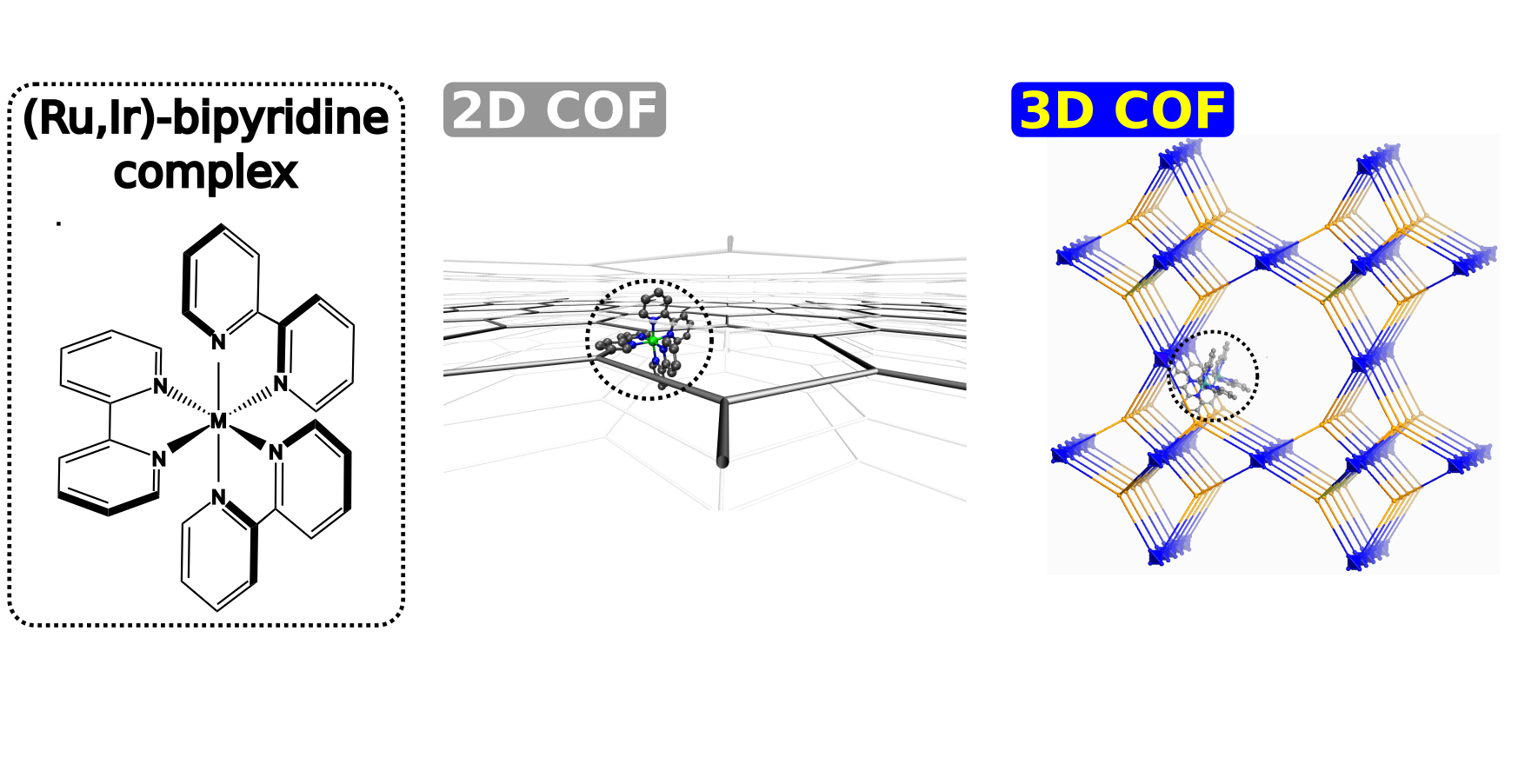
Due to the chemical versatility with which these materials can be synthesized, the number of possible COFs is tremendously high. As a result, it is impossible to experimentally scan all possible candidates for a certain application. Therefore, to exploit the full potential of these extraordinary materials, scientists typically apply both experimental and computational techniques, combining the strengths of both. For example, the resulting diffraction patterns from X-Ray diffraction (XRD) experiments contains information about the geometry and structure of the material and can be compared with the equilibrium geometry predicted by quantum mechanical calculations, while lattice vibrations and the electronic band structure are directly related to various spectroscopic techniques such as Infrared (IR) Absorption Spectroscopy and Ultraviolet-Visible (UV/VIS) Spectroscopy. Furthermore, by means of Far-IR spectroscopy, we can focus on the low-frequency vibrations, which correspond to delocalized degrees of freedom such as shearing.Goal
In this thesis subject, both experimental and computational techniques will be applied to investigate the equilibrium geometries as well as vibrational spectra of various COFs, both 2D and 3D. As such we aim to fully characterize the structure of the COFs, estimating the size of the pores/channels and identifying the vibrational degrees of freedom. Specifically for 2D COFs, the shearing of the sheets will be investigated, both without and with anchored molecules, allowing to elaborate on the impact of anchoring on the mechanical flexibility of the material.
From the experimental point of view, samples will be characterized with X-ray diffraction (XRD), infrared-visible optical absorption spectroscopy and Raman scattering. The XRD pattern provides an easy quality check for the crystallinity of the COFs. Anchoring of metal complexes on the COFs may induces small shifts in the lattice parameters, which may in favorable cases be detectable using XRD. However, very probably anchoring of a metal complex to a COF (or another porous structure) will have an even larger effect on the vibrational and electronic spectra of that metal complex. Therefore, optical absorption spectra extending from the far (low frequency vibrational modes) to the near infrared (electronic transitions) will be measured for COFs on which metal complexes are anchored, and compared with spectra for pure COFs, pure metal complexes. In the far infrared region (30-400 cm-1), the shear vibrational modes of the COFs may be detected as well. The Fourier-transform infrared absorption spectrometer can also use an optically excited spectrum as source. In this way near- to mid-infrared photoluminescence spectra can be recorded and the prospects of investigating metal complex anchored COFs by this method can also be explored.
By means of molecular modeling, one can investigate the structure and vibrational modes on a molecular level. Using quantum mechanical calculations based on Density Functional Theory (DFT), one can compute the geometric and electronic structure of these materials. As a result, we can compute several observables such as the XRD pattern as well as the IR spectrum and compare it with the experimental results. In order to describe the shearing of sheets on a large time and length scale, quantum mechanical calculations may turn out to be computationally too expansive. As a result, one usually applies so-called force fields, which are a mathematical expression for the interaction between the atoms in the molecule (the electrons are not described explicitly) based on simple spring models. The unknown parameters occurring in the energy expression, such as the force constants and rest lengths of the springs, can be estimated either from experimental or quantum mechanical input. Using these force fields, one can perform molecular dynamic simulations that can trace the behavior of covalent organic frameworks through long periods of time, allowing to characterize the dynamical behavior.
This thesis comprises both experiments and computational modeling. Depending on the interest of the student, the focus can be shifted towards one or the other. The materials will be synthesized at COMOC (Center for Ordered Materials, Organometallics and Catalysis, UGent, prof. P. Van Der Voort), hence this research will be performed in close collaboration with this research group.
Aspects
Engineering: The student will apply several experimental and computational techniques to characterize new materials and investigate their potential for various applications.
Physics: The student will use several techniques and interpret their results, which requires a fundamental understanding of quantum mechanics, solid state physics and atomic and molecular physics

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) NANO, MODELLINGKeywordsCovalent organic frameworks, X-Ray Diffraction, Infrared Absorption Spectroscopy, Quantum mechanics, Density functional theory, Force fieldsReferences
[1] A. P. Cote, A I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166-1170
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck