Adsorption energies of isobutene cracking intermediates in zeolites: how accurately can they be determined by first-principle methods
Adsorption energies of isobutene cracking intermediates in zeolites: how accurately can they be determined by first-principle methods
Promotor(en): V. Van Speybroeck, A. Ghysels /17NANO16 / Nanoporous materialsZeolite catalyzed reactions are ubiquitous in chemical industry. Each reaction starts with the formation of a reactive complex, adsorbed to the active site of the framework. (Figure 1) The nature of the active centers in these nanoporous catalysts can be very diverse, varying from Brønsted to Lewis acid sites. A proper estimate of the adsorption enthalpy is a necessary condition for predicting accurate reaction kinetics. Due to the high reactivity of the reaction intermediates at operating conditions, it is hard to acquire the adsorption characteristics experimentally. Therefore, ab initio methods can assist in unravelling the governing intermediates and reaction mechanism. Predicting adsorption energies theoretically also remains a challenging topic. First, the orientation of the adsorbate at the active site is not unique, hence selecting an optimal adsorbate position can be a difficult task. Secondly, an accurate level of theory which can properly account for dispersion interactions should be employed.
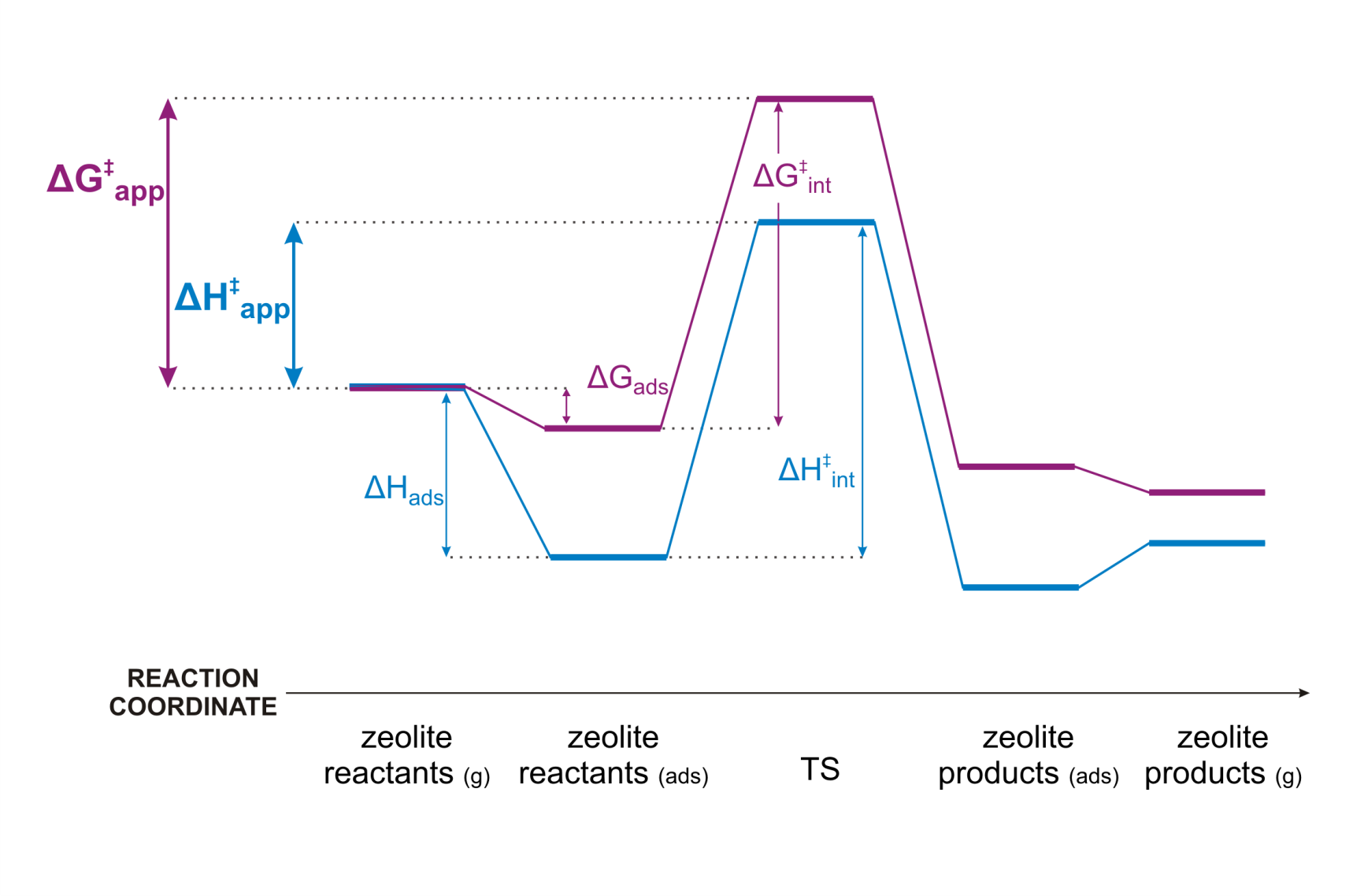
Figure 1. Enthalpy and free energy profile of a zeolite catalyzed reactionAs a case study we consider isobutene cracking in acid zeolites. Upon adsorption of isobutene, three different reactive intermediates can be formed. (Figure 2) The adsorbed reactant can either be a physisorbed π-complex, a chemisorbed carbenium ion or a chemisorbed alkoxide, if isobutene is covalently bound to the framework. Although the topic of isobutene adsorption has been addressed in several theoretical and experimental studies [1-2], there is still no consensus regarding the most stable state. Especially, the existence of stable carbenium ion intermediates is highly controversial. Nevertheless, a good understanding of the nature, stability and dynamic behaviour of catalytic alkene cracking intermediates in is essential to understand the mechanism and kinetics of this process. Qualitatively, it was shown that the tert-butyl carbenium ion becomes significantly more stable than alkoxides or π-complexes at high-temperature cracking conditions. [1] However, a high-level benchmark study using the current state-of-the-art methodologies to investigate (iso)butene adsorption quantitatively is still lacking.
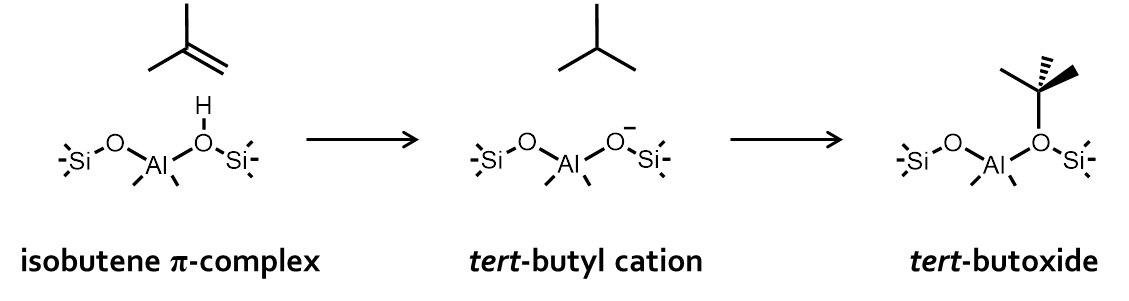
Figure 2. Isobutene adsorption intermediatesObjectives
As a case study we consider isobutene cracking in acid zeolites. Upon adsorption of isobutene, three different reactive intermediates can be formed. (Figure 2) The adsorbed reactant can either be a physisorbed π-complex, a chemisorbed carbenium ion or a chemisorbed alkoxide, if isobutene is covalently bound to the framework. Although the topic of isobutene adsorption has been addressed in several theoretical and experimental studies [1-2], there is still no consensus regarding the most stable state. Especially, the existence of stable carbenium ion intermediates is highly controversial. Nevertheless, a good understanding of the nature, stability and dynamic behaviour of catalytic alkene cracking intermediates in is essential to understand the mechanism and kinetics of this process. Qualitatively, it was shown that the tert-butyl carbenium ion becomes significantly more stable than alkoxides or π-complexes at high-temperature cracking conditions. [1] However, a high-level benchmark study using the current state-of-the-art methodologies to investigate (iso)butene adsorption quantitatively is still lacking.
The main objective of this master thesis is to accurately predict adsorption enthalpies/free energies for isobutene cracking intermediates. To this end, several currently available state-of-the-art techniques will be applied and compared. Ultimately, this will result in a procedure for calculating adsorption energies that can be easily extended to other zeolite catalyzed reactions. First, you will perform a set of static calculations using contemporary hybrid functionals. In a static approach, one explores the energy surface in the immediate vicinity of the selected configuration. This procedure yields a well-defined adsorption enthalpy and free energy at finite temperature; however the solution is not unique. To obtain adsorption enthalpies at finite temperature, thermal energy corrections need to be estimated. These contributions are strongly affected by anharmonicity of the free energy surface, which will be evaluated by the recently developed method of Piccini et al. [3].
Secondly, molecular dynamics simulations on the occurring reactive intermediates will be carried out at operating conditions. With this technique, the free energy surface is explored, taking finite temperature effects, anharmonicity and configurational freedom inherently into account. To resolve discrepancies between dynamic and static techniques, you will perform long MD simulations with a hybrid functional to obtain time averaged adsorption enthalpies. This discussion can also give an answer to the question whether present state-of-the-art techniques are able to predict accurately adsorption energies.
The Center for Molecular Modeling has ample experience in modeling of zeolite catalysis with advanced simulation techniques. The student will be actively coached to get acquainted with the multitude of available techniques to tackle the proposed problem. The proposed topic is challenging and requires technical skills, creativity and chemical insight. The CMM will provide the necessary computational resources for studying this complex system.


- Study programmeMaster of Science in Chemical Engineering [EMCHEM]Recommended coursesMoleculaire modellering van industriële processen, Simulations and Modeling for the Nanoscale
Contact
An Ghysels
Veronique Van Speybroeck
Pieter Cnudde
Kristof De Wispelaere