Abstract
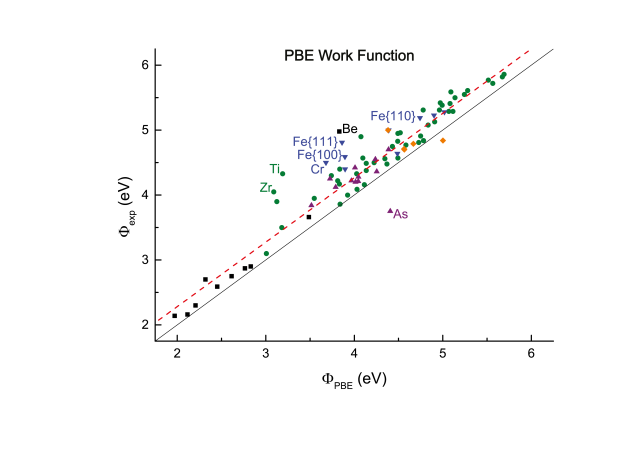
Density-functional theory (DFT) predictions of materials properties are becoming ever more widespread. With increased use comes the demand for estimates of the accuracy of DFT results. In view of the importance of reliable surface properties, this work calculates surface energies and work functions for a large and diverse test set of crystalline solids. They are compared to experimental values by performing a linear regression, which results in a measure of the predictable and material-specific error of the theoretical result. Two of the most prevalent functionals, the local density approximation (LDA) and the Perdew-Burke-Ernzerhof parametrization of the generalized gradient approximation (PBE-GGA), are evaluated and compared. Both LDA and GGA-PBE are found to yield accurate work functions with error bars below 0.3 eV, rivaling the experimental precision. LDA also provides satisfactory estimates for the surface energy with error bars smaller than 10%, but GGA-PBE significantly underestimates the surface energy for materials with a large correlation energy.
 Open Access version available at UGent repository
Open Access version available at UGent repository