R. Declerck
Magnetic linear response properties calculations with the Gaussian and augmented-plane-wave method
Journal of Chemical Physics
131 (1), 014106
2009
A1
Abstract
We introduce a method for the all-electron calculation of the NMR chemical shifts and the EPR g tensor using the Gaussian and augmented-plane-wave method. The presented approach is based on the generalized density functional perturbation theory. The method is validated by comparison with other theoretical methods for a selection of small molecules. We also present two exemplary applications that involve the calculation of the chemical shifts of a hydrated adenine and the g tensor for the E1′ center in α-quartz using a quantum mechanical/molecular mechanical approach.
Evidence for a Grotthuss-like mechanism in the formation of the rhamnose alkoxy radical based on periodic DFT calculations
Radiation Research
169 (1), 8-18
2008
A1
Abstract
Pauwels, E., Declerck, R., Van Speybroeck, V. and Waroquier, M. Evidence for a Grotthuss-Like Mechanism in the Formation of the Rhamnose Alkoxy Radical Based on Periodic DFT Calculations. Radiat. Res. 169, 8-18 (2008). Molecular modeling adopting a periodic approach based on density functional theory (DFT) indicates that a Grotthuss-like mechanism is active in the formation of the radiation-induced alkoxy radical in alpha-l-rhamnose. Starting from an oxidized crystal structure, a hydroxyl proton is transferred along an infinite hydrogen bond chain pervading the entire crystal. The result of this proton shuttling mechanism is a stable radical species dubbed RHop. Only after several reorientations of crystal waters and hydroxyl groups, the more stable radical form RO4 is obtained, which differs in structure from the former by the absence of only one hydrogen bond. Calculations of the energetics associated with the mechanism as well as simulated spectroscopic properties reveal that different variants of the rhamnose alkoxy radical can be observed depending on the temperature of irradiation and consecutive EPR measurement. Cluster calculations on both radical variants provide hyperfine coupling and g tensors that are in good agreement with two independent experimental measurements at different temperatures.
olecular Environment and Temperature Dependence of Hyperfine Interactions in Sugar Crystal Radicals from First Principles
Journal of Physical Chemistry B
112 (5), 1508 -1514
2008
A1
Abstract
The effect of the molecular environment and the temperature dependence of hyperfine parameters in first principles calculations in α-d-glucose and β-d-fructose crystal radicals have been investigated. More specifically, we show how static (0 K) cluster in vacuo hyperfine calculations, commonly used today, deviate from more advanced molecular dynamics calculations at the experimental temperature using periodic boundary conditions. From the latter approach, more useful information can be extracted, allowing us to ascertain the validity of proposed molecular models.
Surface segregation in CuPt alloys by means of an improved modified embedded atom method
Physical Review B
76 (17), 174208
2007
A1
Abstract
We present a procedure to investigate surface structures in CuPt alloys by combining the modified embedded atom method (MEAM) and the cluster expansion method (CEM). While the MEAM provides structural information for the description of extended anisotropic defects, the CEM improves the ability to correctly reproduce the relevant ground state structures in agreement with ab initio data. The procedure is validated with the reproduction of surface energies of pure Cu and Pt, the prediction of TC for order-disorder transitions, the surface and segregation energies in ordered CuPt alloys, and Monte Carlo (MC) simulations of temperature-dependent surface segregation profiles. A complete MEAM-CEM/MC study of the surface segregation in Cu3Pt, CuPt, and CuPt3 alloys is presented, engaging only 11 composition- and volume-independent alloy-specific parameters. Results are critically compared with experimental data from literature and with an independent set of ab initio periodic density functional theory calculations.
First-principles calculations of hyperfine parameters with the Gaussian and augmented-plane-wave method: Application to radicals embedded in a crystalline environment
Physical Review B
74 (24), 245103
2006
A1
Abstract
A method for the calculation of hyperfine parameters in extended systems under periodic boundary conditions is presented, using the Gaussian and augmented-plane-wave density functional method, and implemented in QUICKSTEP. In order to increase the efficiency in larger systems, a hybrid scheme is proposed, in which an all-electron treatment for the nuclei of interest and a pseudopotential approximation for the remaining atoms in the simulation cell are combined. The method is validated first by comparing the hyperfine parameters for a selection of atoms and small molecules (using a supercell technique) with other theoretical methods and experimental data from literature. As a typical example of a periodic system where our hybrid method can be applied, the hyperfine parameters of the well-characterized R2 L-α-alanine derived radical are evaluated, yielding results in excellent agreement with the available experimental data.
First-principles calculation of the EPR g tensor in extended periodic systems
Physical Review B
73 (11), 115113
2006
A1
Abstract
A method for the ab initio prediction of the EPR g tensor for paramagnetic defects in systems under periodic boundary conditions is presented. It is based on density functional theory and the pseudopotential approximation. The formalism is applicable to crystalline and amorphous insulators, as well as to isolated molecules using a supercell technique. The method is validated by comparison with a well-established theoretical approach and experimental data for a series of small isolated molecules. Finally the EPR parameters of an O3− defect in a KCl lattice are evaluated following the new procedure, yielding results in good agreement with experiment and at an attractive computational cost.
Influence of Protein Environment on the Electron Paramagnetic Resonance Properties of Flavoprotein Radicals: A QM/MM Study
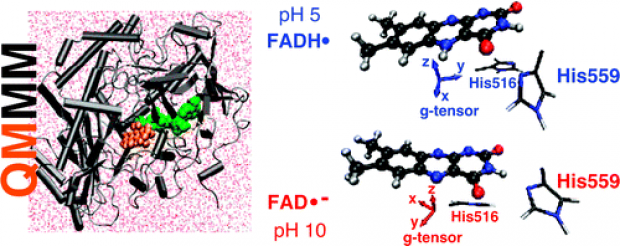
Journal of Physical Chemistry B
114 (49), 16655–16665
2010
A1
Abstract
The neutral and anionic semiquinone radicals of the flavin adenine dinucleotide (FAD) cofactor noncovalently bound in glucose oxidase from A. niger are examined with the aid of QM/MM molecular modeling methods, enabling complete inclusion of the protein environment. Recently, the electron paramagnetic resonance (EPR) characteristics, the anisotropic g tensor and all the significant hyperfine couplings, of these flavoprotein radicals were determined at high resolution (J. Phys. Chem. B 2008, 112, 3568). A striking difference between the neutral and anionic radical forms was found to be a shift in the gy principal value. Within the QM/MM framework, geometry optimization and molecular dynamics simulations are combined with EPR property calculations, employing a recent implementation by some of the authors in the CP2K software package. In this way, spectroscopic characteristics are computed on the fly during the MD simulations of the solvated protein structure, mimicking as best as possible the experimental conditions. The general agreement between calculated and experimental EPR properties is satisfactory and on par with those calculated with other codes (Gaussian 03, ORCA). The protonation state of two histidines (His559 and His516) at the catalytic site of this flavoprotein is found to have a remarkable influence on the isotropic hyperfine coupling of one of the methyl groups on the neutral FAD radical, which is consistent with experimental findings in other flavoproteins (J. Biol. Chem. 2007, 282, 4738). Furthermore, the shift in the gy principal values between the neutral and anionic radicals is well reproduced by QM/MM simulations. Incorporation of at least the nearest protein environment of the cofactor radicals proves to be vital for a correct reproduction, indicating that this shift is a global feature of the protein rather than a local one. In addition, QM/MM techniques are used to make a prediction of relative angles between important spectroscopic principal directions, which are not readily determined by conventional EPR experiments. Significantly, the directions of the gx and the gy components of the g-tensor that lie in the plane of the isoalloxazine moiety are rotated by approximately 59° between the neutral and the anionic radicals.
Insight into the solvation and isomerization of 3-halo-1-azaallylic anions from ab initio metadynamics calculations and NMR experiments
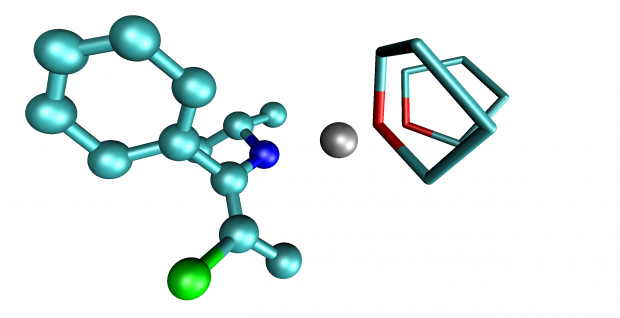
Chemistry - A European Journal
15 (3), 580 - 584
2009
A1

Abstract
Long live theZisomer! The solvation and isomerization properties of lithiated 3-chloro-1-azaallylic anions in tetrahydrofuran are revealed. Extensive and convincing evidence is obtained from state-of-the-art first-principle molecular dynamics and metadynamics simulations in an explicit periodic solvent model, together with detailed NMR experiments.
First-principles calculation of EPR parameters in extended periodic systems
Poster
Conference / event / venue
DFT2005
Geneva, Switzerland
Sunday, 11 September, 2005 to Thursday, 15 September, 2005