Insight into the effects of confined hydrocarbon species on the lifetime of methanol conversion catalysts
Abstract
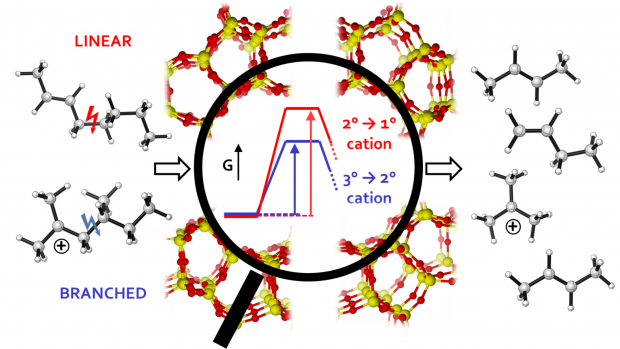
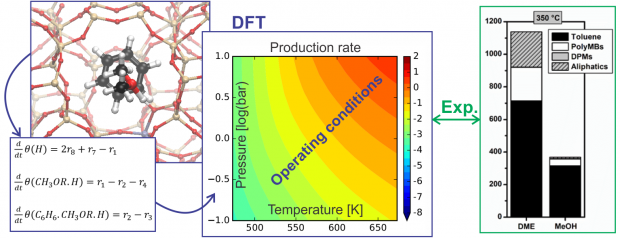
The methanol-to-hydrocarbons reaction refers collectively to a series of important industrial catalytic processes to produce either olefins or gasoline. Mechanistically, methanol conversion proceeds through a ‘pool’ of hydrocarbon species. For the methanol-to-olefins process, these species can be delineated broadly into ‘desired’ lighter olefins and ‘undesired’ heavier fractions that cause deactivation in a matter of hours. The crux in further catalyst optimization is the ability to follow the formation of carbonaceous species during operation. Here, we report the combined results of an operando Kerr-gated Raman spectroscopic study with state-of-the-art operando molecular simulations, which allowed us to follow the formation of hydrocarbon species at various stages of methanol conversion. Polyenes are identified as crucial intermediates towards formation of polycyclic aromatic hydrocarbons, with their fate determined largely by the zeolite topology. Notably, we provide the missing link between active and deactivating species, which allows us to propose potential design rules for future-generation catalysts.
 Open Access version available at
Open Access version available at 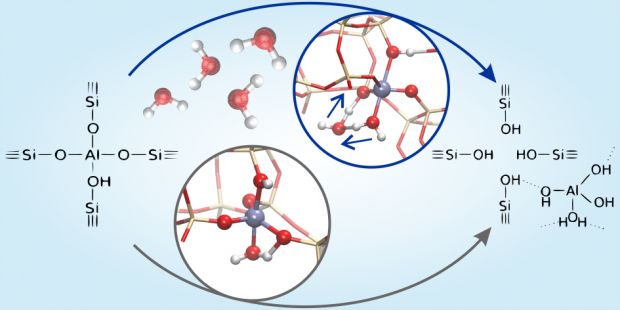
-zeolites is a mobile complex resembling a homogeneous catalyst, tethered by Coulomb interaction to the framework. The complexes are created in situ by coordination of ethene to Ni and lead to second order rate dependence on ethene pressure. Dimerization occurs by the Cossee-Arlman mechanism through transition states having the favoured square-planar coordination of Ni.")